ADVERTISEMENT

The FDA Approval Process: Focus on Accelerated Approval
INTRODUCTION
The United States (U.S.) Food and Drug Administration (FDA) has an important role in the development and approval of new drugs that are introduced to the market. Within the FDA is the Center for Drug Evaluation and Research (CDER), the entity responsible for determining that new drugs are safe and effective for the public. CDER does not test drugs; instead, it is responsible for regulating over-the-counter and prescription medications, biologic therapeutics, and generic medications.1
CDER originated in 1902 when Harvey Wiley, the chief chemist of the Bureau of Chemistry (today’s FDA) created the Drug Laboratory with the plan to help standardize pharmaceuticals and ensure uniform, consistent analytical tests and experiments. The 1906 Federal Food and Drugs Act granted regulatory authority to the administration to achieve its scientific mission.2 This Act also forbade “interstate commerce of mislabeled and adulterated drugs and food.”3 Several other legislative milestones took place over the following century that shaped the FDA into what it is today.
In 1938, Congress passed the Federal Food, Drug, and Cosmetic Act, which created many new provisions related to drug safety. For example, it required manufacturers to demonstrate that new drugs were safe before marketing them to the public. Manufacturers submitted these data to the FDA as part of a new drug application (NDA).3
In 1962, Congress approved the Drug Amendments of 1962, more commonly referred to as the Kefauver-Harris Drug Amendments. These amendments drastically changed the regulatory requirements for drug manufacturers; instead of demonstrating safety alone prior to marketing, they must also prove medications are effective.3
Following approval and marketing of drugs, it was difficult for CDER to determine their safety. As a result, the FDA created two programs: MedWatch and the Drug Safety Oversight Board. MedWatch—a voluntary system for consumers (e.g., physicians and patients) to report adverse events—was first to arrive in 1993, followed by the Drug Safety Oversight Board in 2005 to help advise CDER on drugs after approval.4 Over the span of decades, CDER and the FDA have evolved to improve the safety and efficacy of medications being approved for the public. This is an important consideration, as other programs (e.g., accelerated approval) often prompt questions about medication safety or effectiveness.
Drug Sponsor Application Types
When it comes to new drug development, CDER provides a variety of application types for manufacturers.
For new drugs, an Investigational New Drug Application (IND) precedes an NDA. An IND allows drug manufacturers to move unapproved medications to clinical investigators across state lines, which would otherwise violate federal law. The IND requires manufacturers to perform animal pharmacology and toxicology studies, provide manufacturing information, and detail clinical protocols and investigator information to ensure clinical trial participants will not be subjected to unnecessary risk. After submitting the IND, the sponsor (i.e., the person or company submitting the IND) must wait 30 days before initiating any clinical trials, which gives the FDA a chance to assess the new drug’s safety for potential clinical trial participants.5
Following an IND, drug sponsors can submit an NDA to formally present a new drug to the FDA for U.S. marketing approval. The company includes data from the animal and human trials associated with the IND as part of the NDA.6 The NDA should contain enough information to allow the FDA to determine whether6
- the drug is safe and effective for its proposed use(s)
- the drug’s benefits outweigh its risks
- the drug's proposed labeling (package insert) is appropriate, and what it should contain
- the methods used to manufacture the drug and the controls used to maintain the drug's quality are adequate to preserve the drug's identity, strength, quality, and purity
In the early days of the review process as it exists today, CDER faced challenges with the length of time it took to approve new drugs for marketing. As a result, Congress passed the Prescription Drug User Fee Act (PDUFA) in 1992. This Act established a user fee for drug companies to allow the FDA to increase resources to speed up the review process for new medications. Following the passage of PDUFA, after a manufacturer submits an NDA, the FDA renders a decision regarding the medication’s approval within just 10 months.4
Sponsors use a third application type—the Abbreviated New Drug Application (ANDA)—when they are seeking FDA approval for a generic drug product. These applications are simpler, as they typically do not require animal or human trials. Instead, the sponsor must demonstrate that the medication behaves the same as the innovator drug (i.e., brand name), delivering the same amount of active ingredients into a patient’s bloodstream over the same amount of time.7 In other words, the sponsor must demonstrate bioequivalence between the proposed generic product and the original medication. Following approval, the sponsor can market this generic drug as a safe, effective, and, in most circumstances, lower cost option to the brand name drug.7
After developing the drug and performing the appropriate animal tests and human clinical trials, researchers submit the information for consideration for FDA approval using the applications above. Approval means that CDER analyzed the new drug’s effects and the results show that the drug’s benefits outweigh the risks for the studied population.8
THE DRUG DEVELOPMENT PROCESS
Medications must take a long journey from discovery to patient use. On average, this takes at least 10 years. It is also costly, with estimates suggesting that the entire process can cost an average of $2.6 billion dollars.9 Five major steps occur during the drug development process:
- Discovery and development
- Preclinical research
- Clinical research
- FDA drug review
- FDA post-market drug safety monitoring
Discovery commonly kicks off thanks to new insight into a certain disease state that opens up opportunity for new products (e.g., researchers identify a new potential target to help control or prevent a disease).10 Additionally, researchers may run computational or laboratory tests on thousands of potentially viable molecular compounds in an attempt to identify beneficial effects to treat a disease state.10,11 The development phase then involves identifying all the information needed to start an experiment based on the encouraging compound discovered. Some of this information includes identifying the best dosage, the compound’s potential adverse effects, effectiveness compared to similar alternatives, and the ideal route of administration.10
During preclinical research, scientists conduct testing both in vitro (i.e., within test tubes) and in vivo (i.e., within living organisms) to determine if the newly identified compound causes harm. The FDA requires good laboratory practices (GLP) for preclinical research, which define minimum basic requirements for all aspects of these studies, including the equipment used, operating procedures, and written protocols.12 In all, these first two steps–discovery and development and preclinical research–can take 4 to 7 years to complete.11
When preclinical research concludes, drug developers will submit an IND, and the FDA responds within 30 days with approval to begin clinical trials or a clinical hold to delay or stop the investigation.13 If approved, researchers can then begin clinical trials. Clinical trials are designed based on the specific question researchers are trying to answer. Their protocol must include the selection criteria for participants, the number of projected participants in the study, anticipated study length, if a control group (i.e., a comparator group) will be involved, how investigators will administer the drug to participants, and how researchers plan to collect and review the data.
Within the clinical research step of the drug development process, 4 phases exist11,13:
- Phase 1 includes about 20 to 100 healthy participants with the intention of assessing the new drug’s safety and dosage. This phase may take several months to complete, and about 70% of drugs tested in this phase progress to Phase 2.
- Phase 2 is the “proof-of-concept” stage and involves testing the medication on several hundred participants with the disease or condition for which the drug is intended to gather additional safety data. These studies also allow developers to refine their research questions before initiating larger scale studies in Phase 3, including narrowing down to an optimal dose or patient population. Phase 2 may take up to 2 years to complete, and about one-third of drugs tested in this stage progress to Phase 3.
- Phase 3 includes large scale clinical trials to determine if the medication being studied offers clinical benefit or if any other less common adverse effects occur. The number of participants in these trials is based on how common the disease is but commonly include 300 to 3,000 participants with the condition. This phase may take 1 to 4 years to complete, and only about 25% to 30% of drugs move on to Phase 4.
- Phase 4 (i.e., post-marketing studies) begins following FDA approval and is discussed below.
Upon completion of clinical research, the drug approval process continues to FDA review, which involves the FDA thoroughly assessing preclinical and clinical trial data and making the ultimate decision to approve or reject the medication for use by the public. As previously mentioned, the drug sponsor submits an NDA containing all information that the FDA needs to complete its review. The FDA’s review team can take 6 to 10 months to come to a decision. If the FDA approves the drug, they work with the applicant to develop the product’s labeling to ensure the most appropriate use of that medication. If any questions arise from the NDA, an Advisory Committee will get involved to acquire independent advice and allow the public to make comments.14
Following approval, the drug development process moves into the final step: post-market drug safety monitoring (i.e., monitoring that occurs after the drug is approved), otherwise known as Phase 4 studies. While clinical trials provide safety and efficacy data for the new drug, these trials are only conducted in a small sample of the population. A medication’s true safety and efficacy can take months or years to show, so the FDA monitors medications following approval to ensure no additional concerns arise requiring labeling updates (e.g., new warnings or precautions that the public must be notified about through updated labeling).15
Drug Development Designations
The FDA may assign various drug development designations to a medication during the approval process, including Fast Track, Breakthrough Therapy, and Priority Review.8 These designations (and Accelerated Approval, which will be discussed in more detail later) are independent of each other, and medications can receive any combination of these labels. These various approaches are generally reserved for drugs that are considered the first available treatment for a particular condition or ones that offer a significant benefit compared to other existing therapies for that disease state. Their purpose is to ensure promising new drugs are available to the public as soon as possible once the review team assesses the data.
Fast Track
The purpose of the Fast Track process is to accelerate the development and review of drugs that can treat serious disease states and fill unmet medical needs.8 Serious disease states include those which, left untreated, will progress to a more serious condition. Medications that fill unmet medical needs are ones that either provide a therapeutic option when none currently exist or offer some kind of benefit over existing therapies (i.e., it works significantly better than existing therapies or is likely to help patients avoid serious adverse events or toxicities associated with available therapies). The drug manufacturer must request Fast Track designation. They can do so at any time during the development process, and the FDA will decide on whether to grant Fast Track designation within 60 days of receiving the request. If a drug receives Fast Track designation, the FDA allows for increased communication between the drug manufacturer and the FDA throughout the development process. It also allows for “rolling review” where the drug company can submit sections of its application as they are completed, rather than all at once at the end.16 In 2022, the FDA granted Fast Track status to 32% of novel medications.17
Breakthrough Therapy
The purpose of the Breakthrough Therapy designation is to accelerate the development and review of medications used to treat serious conditions that have preliminary evidence showcasing improvement over existing drugs on one or more clinically significant endpoints. A clinically significant endpoint is defined as one “that measures an effect on irreversible morbidity or mortality, or on symptoms that represent serious consequences of the disease.”18
A drug company submits a request for Breakthrough Therapy designation, typically no later than the end of Phase 2. A medication granted Breakthrough Therapy designation is eligible for all the benefits of Fast Track designation, plus additional FDA guidance on drug development.18 Of all novel medication approvals in 2022, the FDA granted 35% Breakthrough Therapy designation.17
Priority Review
As part of PDUFA, the FDA created 2 categories for review times for NDAs: Standard Review and Priority Review. The FDA completes Standard Reviews within 10 months of receiving the application, while its goal is to take action on medications granted Priority Review within 6 months. The idea is that the FDA will dedicate additional resources and act sooner on applications for medications that offer significant improvement in the safety or effectiveness of existing treatments for serious conditions. Receiving Priority Review designation does not alter required durations for clinical trials, the quality of evidence necessary for approval, or the standard for approval. The FDA typically decides whether an application is given Standard or Priority Review, but drug companies can also request Priority Review.19
Drug manufacturers who develop medications for tropical diseases, rare pediatric diseases, or illnesses related to public health emergencies may be granted a Priority Review Voucher (PRV). This is a kind of “reward” for developing therapies that may not be the most profitable to make. The drug developer can then use a PRV on another medication in development, which may be unrelated to the medication for which the FDA granted the PRV in the first place, or they may sell the PRV to another drug manufacturer.20 The FDA granted Priority Review to 57% of the novel medications approved in 2022.17
Table 1 lists examples of medications recently approved with Fast Track, Breakthrough Therapy, and/or Priority Review designations.
| Table 1. Sample FDA Approvals for Medications Granted Fast Track, Breakthrough Therapy, and/or Priority Review Designations* 17 |
| Brand Name |
Generic Name |
Use |
Fast Track |
Breakthrough Therapy |
Priority Review |
| Amvuttra |
vutrisiran |
Polyneuropathy of hATTR |
✓ |
|
|
| Cibinqo |
abrocitinib |
Refractory atopic dermatitis |
|
✓ |
✓ |
| Voquezna Triple Pak |
vonoprazan, amoxicillin, and clarithromycin (co-packaged) |
Heliobacter pylori infection |
✓ |
|
✓ |
| Sunlenca |
lenacapavir |
Multi-drug resistant HIV infection |
✓ |
✓ |
✓ |
hATTR, hereditary transthyretin-mediated amyloidosis; HIV, human immunodeficiency virus.
*Not an all-inclusive list; used for illustrative purposes only. |
ACCELERATED APPROVAL
Accelerated Approval is a drug designation that differs from others given that it that allows drugs for conditions that fill an unmet medical need to be approved based on a surrogate or intermediate endpoint.21 As opposed to clinical outcomes that directly measure how well a drug impacts the ultimate outcome of interest (e.g., living longer, reducing heart attack risk), surrogate endpoints study an intermediary endpoint that is “reasonably likely to predict a clinical benefit.”
Surrogate endpoints can predict outcomes in many disease states. For example, consider a manufacturer studying a medication to reduce the risk of cardiovascular events. These events take years to develop, so it’s not always feasible to study this as an outcome in clinical studies. Instead, measuring blood pressure lowering as a surrogate outcome is reasonable because it is widely recognized that high blood pressure leads to cardiovascular events.22 Another example would be gout, which results when uric acid accumulates in a joint and causes inflammation and pain. Rather than measuring gout attacks, which take time to develop, a commonly used surrogate endpoint is a reduction in uric acid levels in the blood.22
Studying a surrogate endpoint that is reasonably likely to predict how well a medication will treat or prevent a meaningful clinical outcome may allow patients to receive the medication sooner than the traditional application and review process allows.8
Once the FDA approves a drug through the Accelerated Approval pathway and makes it available to the public, the drug company must complete post-marketing clinical trials to confirm the drug’s clinical benefits. If post-marketing clinical trials do not demonstrate intended benefits, the FDA can withdraw its approval and take the drug off the market.8
Drug companies can also use the Accelerated Approval pathway to repurpose medications currently approved for other uses for additional indications. If a medication has more than one FDA-approved use and the data for an accelerated indication does not pan out (i.e., the benefit suggested by the studies that led to an accelerated approval was not confirmed in subsequent required clinical trials), the FDA will revoke only that indication and the drug can remain on the market. An example of this is the cancer medication, pralsetinib (Gavreto). The FDA revoked pralsetinib’s indication for medullary thyroid cancer in July 2023, but the product remains available on the market for use in patients with non-small cell lung cancer, for which it has full approval, and rearranged during transfection fusion-positive thyroid cancer, for which further study to confirm benefit is ongoing to maintain its accelerated approval.23,24
History of Accelerated Approval
The accelerated approval process has been evolving for many decades. In the 1980s, deaths due to human immunodeficiency virus (HIV) and acquired immunodeficiency syndrome (AIDS) were rising faster than new treatments could be made available. This prompted more discussions and protests for a quicker development and review process for new medications. It took until 1992 to pass legislative changes and establish the Accelerated Approval process, then another 20 years before the FDA Safety and Innovation Act, which reinforced the use of the Accelerated Approval process and surrogate endpoints.25
Since the start of Accelerated Approval thirty years ago, the FDA has granted the designation to more than 250 drugs. Of these accelerated approvals, 125 medications have gone on to receive full FDA approval, 16 have been withdrawn from the market, and 112 remain on the market under accelerated status without a full approval. Figure 1 depicts the outcomes of accelerated approvals over time since the pathway was established in 1992.
| Figure 1. Outcomes of Accelerated Approvals Over Time26 |
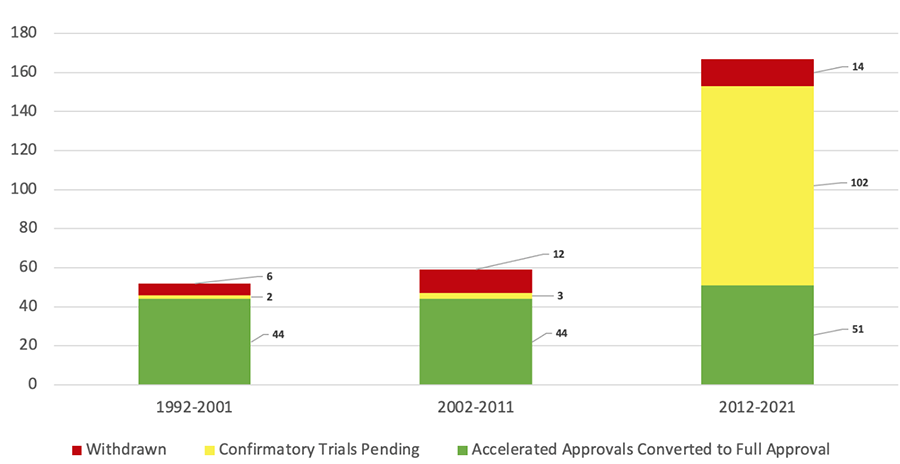 |
In one analysis, the Institute for Clinical and Economic Review identified that 19 medications approved through the Accelerated Approval pathway between 1992 through 2016 had been marketed for a median of 9.5 years without enough evidence to support conversion to full approval.25 The majority (65%) of medications approved through the Accelerated Approval pathway during the first 10 years of the program’s existence were for infectious diseases, which aligns with the historical need for new HIV therapies at the time. More recently, the FDA granted more than 80% of accelerated approvals to oncology (cancer) medications.26 Table 2 lists examples of medications approved through the Accelerated Approval pathway and their current approval status.
| Table 2. Examples of Medications Granted Accelerated Approval24,27-34 |
| Brand Name |
Generic Name |
Accelerated Approval Indication |
Original Approval Date |
Status |
| Infectious Diseases |
| Lampit |
nifurtimox |
Chagas disease |
8/6/2020 |
Verified clinical benefit: 6/2/2023 |
| Sirturo |
bendaquiline |
Pulmonary multidrug resistant TB |
5/27/2022 |
Ongoing study
Projected study completion: 3/31/2022
|
| Sulfamylon |
mafenide acetate |
Bacterial infection under moist dressings over meshed autografts on excised burn wounds |
6/5/1998 |
Withdrawn: 11/30/2022 |
| Cancers |
| Keytruda |
pembrolizumab |
Merkel cell carcinoma |
12/19/2018 |
Verified clinical benefit: 10/12/2023 |
| Elrexfio |
elranatamab-bcmm |
Relapsed or refractory multiple myeloma |
8/14/2023 |
Ongoing study
Projected study completion: 9/30/2026
|
| Gavreto |
pralsetinib |
Medullary thyroid cancer |
12/1/2020 |
Withdrawn: 7/20/2023 |
| Non-Malignant Hematologic, Neurologic, and Other Disorders |
| Leqembi |
lecanemab-irmb |
Alzheimer’s disease |
1/6/2023 |
Verified clinical benefit: 7/6/2023 |
| Qalsody |
tofersen |
ALS in adults with a mutation in the SOD1 gene |
4/25/2023 |
Ongoing study
Projected study completion: 6/30/2028
|
| Makena |
hydroxyprogesterone caproate |
Reduce the risk of preterm birth |
2/3/2011 |
Withdrawn: 4/6/2023 |
| ALS, amyotrophic lateral sclerosis; SOD1, superoxide dismutase 1; TB, tuberculosis. |
Mixed viewpoints exist regarding whether the Accelerated Approval process is a success or failure. On one hand, more medications approved through the Accelerated Approval pathway have received full approvals than have been withdrawn. Conversely, some believe the approval process is too generous and that post-marketing confirmatory trials move too slowly. Additionally, when manufacturers submit study results to the FDA, only 55% show data that verify the benefits on which approval had initially been granted.25 The SIDEBAR outlines a case study reflecting these challenges.
| SIDEBAR: Case Study: Hydroxyprogesterone Caproate |
The FDA originally approved hydroxyprogesterone caproate injection in 2011 through the Accelerated Approval pathway to reduce the risk of preterm birth in women pregnant with one baby who have a history of spontaneous preterm birth.35 The drug’s approval was based on findings from Trial 002, which showed some benefit on the rate of recurrent preterm birth (a surrogate endpoint for improved neonatal outcomes). However, some methodologic issues with the study’s statistical analysis could have increased the risk of false positive results.36 Rather than delaying the medication’s entry to the market, the FDA recognized the potential benefit, approved the medication through the Accelerated Approval pathway, and required confirmatory trials to verify the proposed clinical benefit.36
Unfortunately, the confirmatory trial (Trial 003) found conflicting results on the surrogate endpoint studied in Trial 002, preterm birth. Additionally, Trial 003 also demonstrated a lack of effect on neonatal outcomes, indicating that preterm birth was not an effective surrogate endpoint.36 As a result, in 2020, approximately nine years after its original approval, CDER proposed withdrawing the drug’s approval and ultimately withdrew the approval of both brand and generic hydroxyprogesterone caproate injection in April 2023.35
This example shows that the Accelerated Approval process has its challenges and that there are times when follow-up studies do not confirm safety and effectiveness. Early clinical trials of a drug can indicate the potential for clinical benefit based on a surrogate endpoint. However, proper analysis of these trials can help prevent the approval of drugs with limited or no therapeutic value. |
Benefits and Challenges of the Accelerated Approval Process
The Accelerated Approval process provides several notable benefits, both for society and for drug manufacturers. The original intent of the Accelerated Approval pathway highlights perhaps the greatest societal benefit of this program: to allow medications to be made available to the public quicker than through traditional means.
A study conducted by the Partnership for Health Analytic Research showcased the benefit to life that medications approved through the Accelerated Approval pathway could have. Included in this analysis were two drugs, alectinib and pemetrexed, which received Accelerated Approval for the treatment of patients with non-small cell lung cancer that had progressed or did not respond to prior therapy. The study estimated that approving these medications on an abbreviated timeline contributed to patients living nearly 200,000 additional years versus what would have been expected without treatment.37 The FDA approved ritonavir through the Accelerated Approval pathway for HIV, helping more than 100,000 patients live longer and experience fewer AIDS-related complications.37 These examples demonstrate that the Accelerated Approval process has been beneficial in treatment of multiple disease states, improving the quality and length of life for patients due to these drugs’ early availability.
Another benefit of the Accelerated Approval process is creating a financial incentive for drug makers.25 This pathway allows manufacturers to commercialize medications sooner, which allows them to generate sales revenue while still conducting confirmatory trials. This encourages drug manufacturers to seek treatments for disease states with limited therapeutic options, or as defined by the purpose of the program, “serious conditions that fill an unmet medical need.”21
Alongside the benefits, Accelerated Approval is also associated with challenges. One of these involves using unvalidated surrogate endpoints under the assumption that they can reasonably predict a clinically meaningful outcome. Experts anticipate that improving surrogate endpoints will lead to favorable downstream therapeutic effects. However, when data are scarce to connect a surrogate endpoint to the outcome of interest, it may mean that the medication does not work as well as was expected and could also lead to devastating consequences.
One such example includes the surrogate endpoint of ventricular arrhythmia (abnormal heart rhythm that leads to ineffective pumping). Since ventricular arrhythmia can lead to serious cardiac complications and death, researchers expected that controlling ventricular arrhythmia would reduce mortality in patients who previously had a heart attack. Based on this assumption, the FDA approved 3 medications for this indication based on the surrogate endpoint of ventricular arrhythmia suppression. Following approval and use of these therapies in many patients, a clinical trial unexpectedly found that medications that suppress ventricular arrhythmias actually increased the risk of death three-fold compared with placebo. Researchers estimate that at least 50,000 excess deaths occurred before this study was published.25 This example demonstrates that using unvalidated surrogate endpoints for Accelerated Approval can have tragic consequences.
Another challenge with Accelerated Approval is delayed and/or inadequate confirmatory trials. Post-marketing trial performance and reporting often falls short for multiple reasons, including the fact it is challenging to design a clinical trial with a medication that has received FDA approval, regardless of an Accelerated Approval designation. The medication is no longer considered “investigational” and for that reason may be used by patients outside of clinical studies. It becomes challenging for manufacturers to enroll patients in a clinical study where they may receive a placebo when they have the option of a health care provider prescribing the medication. Without these confirmatory trials, drugs approved through the Accelerated Approval process may remain on the market for years without full confirmation that they are safe and/or effective.25
A related challenge is that drug sponsors are not financially incentivized to rush to conduct these confirmatory studies. Before the confirmatory studies are completed, the therapy remains on the market to be sold and distributed to patients. If the results of a confirmatory study are unfavorable (i.e., do not confirm medication benefit or safety), the millions or billions of dollars invested in developing and marketing that medication may be lost. If a manufacturer is directly competing with other therapies with established clinical benefit and full FDA approval, the financial incentives align more closely.25 Additionally, drug sponsors are ethically incentivized to “do the right thing” and ensure their medications are bringing value to the patients that use them.
Many medications approved through the Accelerated Approval pathway are high-cost specialty medications used to treat orphan diseases (i.e., conditions that affect less than 200,000 people in the U.S.). To put it into perspective, medications approved through the Accelerated Approval pathway in 2020 ranged between approximately $13,400 and $22,600 per month, with one outlier costing approximately $54,000 per month. This was comparable to specialty medications that had received full approval.25 The concern is that the clinical benefit, and therefore the actual value of medications receiving Accelerated Approval, will not be proven through the required confirmatory clinical studies before these high prices are set.
Future of Accelerated Approval
Given the benefits and drawbacks of the Accelerated Approval review process, regulatory authorities must strike a balance to positively impact patients’ health while ensuring medication safety. Legislative reforms aim to find this balance and strengthen the process as a whole.
In response to concerns that ineffective drugs are not removed from the market in a timely enough manner, Congress enacted reform as part of the Consolidated Appropriations Act of 2023. This Act contains the Food and Drug Omnibus Reform Act (FDORA) of 2022, which included section 3210, entitled “Modernizing Accelerated Approval.” This law requires drug companies to specify conditions for post-approval trials no later than the date of Accelerated Approval. Drug companies with medications approved through this process must also submit a progress report on the post-confirmatory trials every 6 months.38 Other changes in this Act include clarifying an expedited process to remove medications previously granted Accelerated Approval and a requirement for the FDA to develop the formation of an intra-agency coordinating council to consistently maintain appropriate use of the Accelerated Approval process.38,39 The goal of reform like this is for the Accelerated Approval process to continue to develop in such a way that patients can continue to safely benefit from this pathway with fewer drawbacks and challenges.
Conclusion
The FDA approval process has changed dramatically over the years and will continue to evolve with further government reform. The introduction of various approval pathways—including the Accelerated Approval process—has given patients earlier access to life-changing therapies, though not without risk. Pharmacy technicians should remain up to date on drug development and FDA approval processes to appreciate the rigor that goes into bringing medications to the market. Understanding the nuances between the various approval pathways and the benefits and drawbacks of the Accelerated Approval process can help pharmacy technicians discuss these concepts with patients should questions arise.
REFERENCES
- U.S. Food and Drug Administration. Center for Drug Evaluation and Research | CDER. Updated August 14, 2023. Accessed December 17, 2023. https://www.fda.gov/about-fda/fda-organization/center-drug-evaluation-and-research-cder
- Swann JP. FDA’s origin. U.S. Food and Drug Administration. Updated February 1, 2018. Accessed December 17, 2023. https://www.fda.gov/about-fda/changes-science-law-and-regulatory-authorities/fdas-origin
- U.S. Food and Drug Administration. A brief history of the Center for Drug Evaluation and Research. Updated January 31, 2018. Accessed December 17, 2023. https://www.fda.gov/about-fda/fda-history-exhibits/brief-history-center-drug-evaluation-and-research
- Meadows M. Promoting safe & effective drugs for 100 years. U.S. Food and Drug Administration. Updated April 23, 2019. Accessed December 17, 2023. https://www.fda.gov/about-fda/histories-product-regulation/promoting-safe-effective-drugs-100-years
- U.S. Food and Drug Administration. Investigational New Drug (IND) application. Updated July 20, 2022. Accessed December 17, 2023. https://www.fda.gov/drugs/types-applications/investigational-new-drug-ind-application
- U.S. Food and Drug Administration. New Drug Application (NDA). Updated January 21, 2022. Accessed December 17, 2023. https://www.fda.gov/drugs/types-applications/new-drug-application-nda
- U.S. Food and Drug Administration. Abbreviated New Drug Application (ANDA). Updated December 16, 2022. Accessed December 17, 2023. https://www.fda.gov/drugs/types-applications/abbreviated-new-drug-application-anda
- U.S. Food and Drug Administration. Development & approval process. Updated August 8, 2022. Accessed December 17, 2023. https://www.fda.gov/drugs/development-approval-process-drugs
- Pharmaceutical Research and Manufacturers of America (PhRMA). Clinical trials. Updated May 2022. Accessed December 17, 2023. https://phrma.org/en/policy-issues/Research-and-Development-Policy-Framework/Clinical-Trials
- U.S. Food and Drug Administration. Step 1: discovery and development. Updated January 4, 2018. Accessed December 17, 2023. https://www.fda.gov/patients/drug-development-process/step-1-discovery-and-development
- BioStock. Drug development – the four phases. January 2, 2023. Accessed December 17, 2023. Available at: https://www.biostock.se/en/2023/01/drug-development-the-four-phases/
- U.S. Food and Drug Administration. Step 2: preclinical research. Updated January 4, 2018. Accessed December 17, 2023. https://www.fda.gov/patients/drug-development-process/step-2-preclinical-research
- U.S. Food and Drug Administration. Step 3: clinical research. Updated January 4, 2018. Accessed December 17, 2023. https://www.fda.gov/patients/drug-development-process/step-3-clinical-research
- U.S. Food and Drug Administration. Step 4: FDA drug review. Updated January 4, 2018. Accessed December 17, 2023. https://www.fda.gov/patients/drug-development-process/step-4-fda-drug-review
- U.S. Food and Drug Administration. Step 5: FDA post-market drug safety monitoring. Updated January 4, 2018. Accessed December 17, 2023. https://www.fda.gov/patients/drug-development-process/step-5-fda-post-market-drug-safety-monitoring
- U.S. Food and Drug Administration. Fast track. Updated January 4, 2018. Accessed December 17, 2023. https://www.fda.gov/patients/fast-track-breakthrough-therapy-accelerated-approval-priority-review/fast-track
- U.S. Food and Drug Administration. Advancing health through innovation: New drug therapy approvals 2022. January 2023. Accessed December 17, 2023. https://www.fda.gov/media/164429/download?attachment
- U.S. Food and Drug Administration. Breakthrough Therapy. Updated January 4, 2018. Accessed December 17, 2023. https://www.fda.gov/patients/fast-track-breakthrough-therapy-accelerated-approval-priority-review/breakthrough-therapy
- U.S. Food and Drug Administration. Priority Review. Updated January 4, 2018. Accessed December 17, 2023. https://www.fda.gov/patients/fast-track-breakthrough-therapy-accelerated-approval-priority-review/priority-review
- U.S. Government Accountability Office. Drug development: FDA’s Priority Review voucher programs. January 31, 2020. Accessed December 17, 2023. https://www.gao.gov/products/gao-20-251
- U.S. Food and Drug Administration. Accelerated approval. Updated February 24, 2023. Accessed December 17, 2023. https://www.fda.gov/patients/fast-track-breakthrough-therapy-accelerated-approval-priority-review/accelerated-approval
- U.S. Food and Drug Administration. Table of surrogate endpoints that were the basis of drug approval or licensure. Updated February 28, 2022. Accessed December 17, 2023. https://www.fda.gov/drugs/development-resources/table-surrogate-endpoints-were-basis-drug-approval-or-licensure
- Gavreto (pralsetinib). Prescribing information. Genentech, Inc.; August 2023.
- U.S. Food and Drug Administration. Withdrawn | cancer accelerated approvals. Updated July 25, 2023. Accessed December 17, 2023. https://www.fda.gov/drugs/resources-information-approved-drugs/withdrawn-cancer-accelerated-approvals
- Kaltenboeck A, Mehlman A, Pearson SD. Strengthening the Accelerated Approval pathway: An analysis of potential policy reforms and their impact on uncertainty, access, innovation, and costs. Institute for Clinical and Economic Review. April 26, 2021. Accessed December 17, 2023. https://icer.org/wp-content/uploads/2021/04/Strengthening-the-Accelerated-Approval-Pathway-_-ICER-White-Paper-_-April-2021.pdf
- Beakes-Read G, Neisser M, Frey P, Guarducci M. Analysis of FDA's Accelerated Approval program performance December 1992-December 2021. Ther Innov Regul Sci. 2022;56(5):698-703. doi:10.1007/s43441-022-00430-z
- U.S. Food and Drug Administration. Ongoing | infectious disease accelerated approvals (excluding vaccines). Updated August 23, 2023. Accessed December 17, 2023. https://www.fda.gov/drugs/accelerated-approval-program/ongoing-infectious-disease-accelerated-approvals-excluding-vaccines
- U.S. Food and Drug Administration. Verified clinical benefit | infectious disease accelerated approvals (excluding vaccines). Updated August 24, 2023. Accessed December 17, 2023. https://www.fda.gov/drugs/accelerated-approval-program/verified-clinical-benefit-infectious-disease-accelerated-approvals-excluding-vaccines
- U.S. Food and Drug Administration. Withdrawn | infectious disease accelerated approvals (excluding vaccines). Updated February 16, 2023. Accessed December 17, 2023. https://www.fda.gov/drugs/accelerated-approval-program/withdrawn-infectious-disease-accelerated-approvals
- U.S. Food and Drug Administration. Verified clinical benefit | non-malignant hematological, neurological, and other disorder indications accelerated approvals. Updated October 31, 2023. Accessed December 17, 2023. https://www.fda.gov/drugs/accelerated-approval-program/verified-clinical-benefit-non-malignant-hematological-neurological-and-other-disorder-indications
- U.S. Food and Drug Administration. Ongoing | non-malignant hematological, neurological, and other disorder indications accelerated approvals. Updated October 31, 2023. Accessed December 17, 2023. https://www.fda.gov/drugs/accelerated-approval-program/ongoing-non-malignant-hematological-neurological-and-other-disorder-indications-accelerated
- U.S. Food and Drug Administration. Withdrawn | non-malignant hematological, neurological, and other disorder indications accelerated approvals. Updated August 24, 2023. Accessed December 17, 2023. https://www.fda.gov/drugs/accelerated-approval-program/withdrawn-non-malignant-hematological-neurological-and-other-disorder-indications-accelerated
- U.S. Food and Drug Administration. Verified clinical benefit | cancer accelerated approvals. Updated December 15, 2023. Accessed December 17, 2023. https://www.fda.gov/drugs/resources-information-approved-drugs/verified-clinical-benefit-cancer-accelerated-approvals
- U.S. Food and Drug Administration. Ongoing | cancer accelerated approvals. Updated July 25, 2023. Accessed December 5, 2023. https://www.fda.gov/drugs/resources-information-approved-drugs/ongoing-cancer-accelerated-approvals
- U.S. Food and Drug Administration. FDA Commissioner and chief scientist announce decision to withdraw approval of Makena. April 6, 2023. Accessed December 17, 2023. https://www.fda.gov/news-events/press-announcements/fda-commissioner-and-chief-scientist-announce-decision-withdraw-approval-makena
- Chang CY, Nguyen CP, Wesley B, et al. Withdrawing approval of Makena - a proposal from the FDA Center for Drug Evaluation and Research. N Engl J Med. 2020;383(24):e131. doi:10.1056/NEJMp2031055
- Ortendahl JD, Broder MS. Clinical benefits of accelerated approval. Partnership for Health Analytic Research. July 26, 2022. Accessed December 16, 2023. https://www.pharllc.com/wp-content/uploads/2022/07/Clinical-Benefits-of-Accelerated-Approval-Brief.pdf
- Congress.gov. H.R.2617 – Consolidated Appropriations Act, 2023. Accessed December 16, 2023. https://www.congress.gov/bill/117th-congress/house-bill/2617/text
- Cooley. FDORA changes to the FDA Accelerated Approval program. January 31, 2023. Accessed December 16, 2023. https://www.cooley.com/news/insight/2023/2023-01-31-fdora-changes-to-the-fda-accelerated-approval-program
Back to Top