
Expired activity
Please go to the PowerPak
homepage and select a course.
Alzheimer's Disease: Addressing the Underlying Mechanisms of Preclinical Disease and Mild Cognitive Impairment (Monograph)
UNMET NEEDS IN ALZHEIMER’S DISEASE
Alzheimer's disease (AD) is a growing epidemic and is considered one of the "greatest medical challenges that face this century."1 Although increasing in prevalence, current treatment options are limited, primarily focused on symptomatic control of mild-to-moderate clinical symptoms that manifest after a long period of clinically silent neurodegeneration.1 AD is an irreversible, progressive cognitive disorder that impairs memory, orientation, and functional capacity beyond what is considered part of the normal aging process.2 It is the most common cause of dementia, accounting for 60% to 80% of all cases worldwide.3 Epidemiology studies have found that the prevalence and incidence of patients living with AD increase exponentially from the age of 65 years onward, reaching a prevalence rate of 22.5% in those older than 85 years. An estimated 50 million people live with AD-associated dementia worldwide.2 In 2021 in the United States (US) alone, an estimated 6 million Americans aged 65 and older were living with AD, and this number is projected to double by the year 2050.4
Although the risk of developing AD is multifactorial, age is considered the most significant risk factor and the best predictor of developing the disease. By 2030, the proportion of the US population 65 years and older is forecasted to grow dramatically to an estimated 72 million older Americans. These older adults will make up approximately 20% of the total population (up from 13% in 2010), placing an even greater proportion of the US population at risk of developing AD. Using data from the Framingham Study, the estimated lifetime risk of a 65-year-old woman without dementia developing AD dementia during her lifetime is 21%, compared with a 12% chance for a man of the same age.5
Alzheimer’s disease is the sixth leading cause of death in the US, and among the subset of the population aged 65 years and older, AD is the fifth leading cause of death.5 From 2000 to 2018, AD-related deaths increased by approximately 146%. This contrasts with the number of deaths attributed to other major diseases.3 For example, between 2000 and 2010, the proportion of deaths due to heart disease decreased by 16% and stroke-related deaths decreased by 23%.5 In addition to being a leading cause of mortality, AD results in significant morbidity and reduced quality of life.2
As the number of elderly adults increases, so do the projected economic costs of treating and caring for patients living with AD. A trajectory report published by the Alzheimer's Association in 2015 predicts that a treatment introduced in 2025 that delays the onset of AD by 5 years would immediately reduce the number of patients affected by the disease.6 In the first 5 years alone, treatment would decrease the number of Americans aged 65 and older living with the disease from 8.2 million to 5.8 million, resulting in an estimated $83 billion savings for payers and a $20 billion out-of-pocket costs saving for patients.6 In 2013, more than 15 million family members and other unpaid caregivers provided an estimated 17.7 billion hours of unpaid care to people living with AD and other dementias, a contribution to the nation valued at over $220 billion.5
Patient Scenario
AJ is a pleasant 75-year-old lady that lives independently in the community. She is a regular customer at the pharmacy and has a history of diabetes mellitus and hypertension. Today, she seems quieter than usual and proceeds to tell you that her kids made her go to the doctor to get her memory checked because she keeps forgetting things. After lab work, memory test, and a brain scan, her doctor told her she had mild cognitive impairment (MCI) due to AD and gave her a new prescription for donepezil. She is afraid of being sent to live at a nursing home and wants to know how long it will take before her new medicine fixes her MCI.
Currently, there is no “fix” or cure for AD. Before the approval of the first disease-modifying therapy in June of 2021, there were no treatments available in the US to slow or prevent AD-related neurodegeneration, only treatments to temporarily improve cognition and neuropsychiatric symptoms.3 Symptomatic treatments include 3 cholinesterase inhibitors (donepezil, galantamine, and rivastigmine) and 1 noncompetitive N-methyl-D-aspartate (NMDA) receptor antagonist (memantine). Cholinesterase inhibitors enhance cholinergic neurotransmission by blocking the enzyme acetylcholinesterase, responsible for the breakdown of acetylcholine, a neurotransmitter associated with memory.7 An overactive glutaminergic system in the brain is thought to be involved in AD-related neurotoxicity. Memantine blocks the NMDA-receptor subtype of glutamate receptors preventing overactivation of glutamine receptors.8 No FDA-approved medications are approved to treat behavioral and psychiatric symptoms that may develop in the later stages of AD.4 A detailed discussion of symptomatic AD treatments is beyond the scope of this educational activity.
There is an increasing need for safe and effective diseasemodifying therapies to prevent or slow AD progression. After factoring in the direct health and long-term care costs for people living with AD and other dementias, AD is one of the costliest conditions in the US.9 In addition, the quality of life of patients living with AD worsens as the disease progresses, negatively impacting both patients and their families. On average, a person with AD will spend more years (40% of the total number of years with AD) in the most severe stage of the disease than in any other stage and will spend most of this time in a nursing home. Approximately two-thirds of patients who die of dementia do so in nursing homes compared to 20% of people with cancer and 28% of people dying from other conditions.5
The lack of available disease-modifying therapies and increasing demand for innovative new treatments have motivated the research for much-needed therapeutic alternatives directed at amyloid-beta (Aβ) pathology and tau pathology.2 The first and currently only FDA-approved medication to address this unmet need is aducanumab. Aducanumab is a human, immunoglobulin gamma 1 (IgG1) monoclonal antibody directed against aggregated forms of Aβ.3 It was studied in and approved to treat patients with MCI, or the mild dementia stage of AD.3,10 Other anti-Aβ monoclonal antibodies in late-stage clinical development include lecanemab, donanemab, and gantenerumab, all of which target aggregated forms of Aβ.10
AJ’s fear of ending up at a nursing home is justified as the average person with AD spends 40% of the total number of years with AD in a nursing home. Currently, there is no “fix” or cure for AD. Most available treatment options are for symptomatic control of patients with mild-to-moderate disease and do not slow or prevent the underlying pathological changes that result in progressive and irreversible neurodegeneration associated with AD. To help ease AJ’s concerns, the pharmacist can discuss the approval of aducanumab. Although not a cure for AD, it is an option for select patients with MCI to slow AD progression.
REVIEW OF AD PATHOLOGY
AD is the most common subtype of dementia, followed by vascular dementia and other neurodegenerative dementias, such as dementia due to Lewy bodies, Parkinsonism-dementia complex, and frontotemporal dementia.11 Different subtypes of dementia are characterized by a clinical picture with common symptoms that differ in age at onset, clinical presentations, clinical course, and associated symptoms. Autopsy studies have found that most people with AD neuropathology also have pathological changes consistent with a second cause of dementia. Autopsy findings from a study of adults clinically diagnosed with AD (N = 477) found that 3% had evidence of AD-related neuropathological only, 15% had evidence of neuropathological consistent with a different type of dementia, and 82% had changes of at least 1 other type of dementia plus AD-related neuropathology.4
The underlying neurodegenerative process of AD begins decades before symptoms emerge.4 Patients remain asymptomatic during the initial preclinical stage of the disease. However, they do have evidence of accumulating neurotoxicity and synaptic dysfunction in brain regions involved in memory, including the entorhinal cortex and hippocampus, that can be detected using biomarkers (measurable indicators of disease). As the disease progresses, areas in the cerebral cortex responsible for language, reasoning, and social behavior are destroyed.12 The preclinical phase of AD has been compared to the preclinical phase of other chronic diseases such as diabetes mellitus, hypertension, cancer, and osteoporosis, all of which can be detected early using biomarkers. Early identification and initiation of safe and effective treatments for these conditions can prevent or slow disease progression.13
After years of accumulating neurotoxicity, patients living with AD start to notice subtle changes in their memory. When the patient or their family have concerns accompany these changes, patients move into the next phase of the disease called mild cognitive impairment (MCI). For a clinical diagnosis of MCI, patients must be concerned about their declining cognition, and the change should be greater than expected for the patient's age and level of education. Patients may have mild functional impairment during this second phase of the disease for complex tasks, but their symptoms are not severe enough to interfere with daily activities. As the disease progresses and symptoms become more noticeable and start to impact the patient’s ability to perform daily activities, the patient meets the criteria for the final stage of the disease: dementia due to AD (FIGURE 1).14
| Figure 1. Alzheimer’s Disease Stages14 |
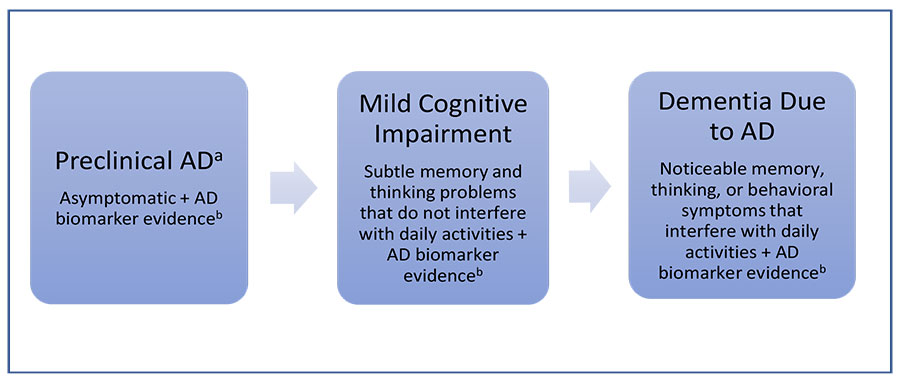 |
Abbreviations: AD, Alzheimer’s disease; Aβ, amyloid-beta; PET, positron emission tomography.
aNot all people with evidence of AD-related brain changes progress to mild cognitive impairment or dementia due to AD. For example, some people have Aβ plaques at death but do not have memory or thinking problems while alive.
bExamples of AD biomarkers include abnormal levels of Aβ as shown on PET scans, an analysis of cerebrospinal fluid, and decreased metabolism of glucose as shown on PET scans. |
AD-related neuropathological changes include the formation of Aβ plaques and phosphorylated tau (p-tau) neurofibrillary tangles (NFTs).15 Aβ peptides are formed from the breakdown of amyloid precursor protein (APP), a large protein, via enzymatic cleavage by the β- and γ-secretases enzymes.16 Aβ is present in both healthy and AD human brain tissue. Significant levels of Aβ accumulation have been detected in approximately 30% of normal older individuals without clinical evidence of AD.15 Multiple isoforms of Aβ exist that differ based on the number of amino acid residues. The Aβ42 isoform is considered the more toxic isoform, and several studies have found that high levels can result in neuronal degeneration and synapse dysfunction. An accumulation and increase in Aβ42 associated with aging have been observed before the appearance of amyloid plaques.2
Microtubules are structures responsible for guiding nutrients and molecules from the cell body to the axon and dendrites. Tau proteins help stabilize the microtubules of neurons. In AD, abnormal hyperphosphorylated tau and aggregated tau can lead to the formation of filaments within neurons. These filaments condense to form NFTs, which block the neuron's transport system and lead to neurodegeneration.12,17
Chronic neuroinflammation also plays a significant role in the neurodegenerative process of AD.2 presence of Aβ and abnormal tau activate microglia.4,12 Microglial cells are specialized glial cells that remove damaged neurons and infections and are essential for maintaining the health of brain cells.4 In AD, microglia fail to remove waste, debris, and protein collections, including Aβ plaques.4,12 Microglia represent the first line of defense for brain cells, and their activation has been observed when the levels of Aβ increase without necessarily forming Aβ plaques. When Aβ plaques appear, microglial cells move towards and surround the plaques to prevent further growth but are not believed to have phagocytic activity against the plaques. The microglia remain active from the beginning of AD, establishing a pattern of proinflammatory cytokines and neurotoxicity which contributes to and correlates with the neuronal death observed in AD.2
The amyloid cascade hypothesis is the prevailing research theory behind the neuropathology of AD. The original theory hypothesizes that the formation of Aβ plaques is a critical early trigger that precedes and accelerates the formation of tau NFTs. However, more recent studies suggest that the oligomeric forms of Aβ peptides (small aggregates of 2 to 12 peptides) are more toxic to neurons than the actual Aβ plaques.15,18
Risk-Reduction Strategies
After learning that there are no treatments available to cure her MCI, AJ wants to know what she can do to prevent her MCI from progressing to dementia due to AD. Thus, she asks the pharmacist, what lifestyle changes can she make to reduce her risk?
Multiple AD risk-reduction strategies have been identified that could potentially prevent or slow disease progression. Risk-reducing strategies can be classified as unmodifiable or modifiable. Unmodifiable risks include advancing age, female gender, and genetic mutations.11 More than 20 genes have been identified that could potentially affect a person's risk of developing dementia. However, genetic causes account for less than 1% of all AD subtypes.11,19 The gene associated with the greatest risk of developing AD is the apolipoprotein E (APoE) gene, with the APoE4 variant posing the highest risk.4
Twelve modifiable risk-reduction strategies have been identified that can prevent or delay approximately up to 40% of dementias. Modifiable risk-reduction strategies can be divided into those that reduce AD neuropathology and those that increase and maintain cognitive function (TABLE 1).4,20
| Table 1. Modifiable Risk Strategies to Potentially Prevent or Delay Dementia20 |
| Risk-Reduction Strategies |
Reduce AD
Neuropathologya |
Increase and Maintain
Cognitive Function |
| Control diabetes |
Yes |
No |
| Treat hypertension |
Yes |
No |
| Prevent head injuries |
Yes |
No |
| Smoking cessation |
Yes |
No |
| Reduce air pollution |
Yes |
No |
| Reduce midlife obesity |
Yes |
No |
| Frequent exercise |
Yes |
Yes |
| Prevent depression |
Yes |
Yes |
| Avoid excessive alcohol use |
Yes |
Yes |
| Treat hearing impairment |
No |
Yes |
| Frequent social interaction |
No |
Yes |
| High level of education |
No |
Yes |
Abbreviation: AD, Alzheimer’s disease.
aAmyloid- or tau-mediated or neuroinflammatory. |
After reviewing AJ’s refill history for medication adherence and noticing that she fills her diabetes and blood pressure medications regularly, the pharmacist can congratulate her on taking her medications as prescribed. The pharmacist can explain that although we do not know the actual cause of AD, 12 potentially modifiable risk factors have been found to potentially prevent or delay approximately 40% of AD cases. For AJ, potential risk-reduction strategies include continued control of her diabetes and blood pressure, maintaining a healthy weight, avoiding alcohol and smoking, frequent social interaction, frequent exercise, and making sure she has no hearing problems.
WHAT’S NEW IN AD RESEARCH
Biomarkers used in AD to measure disease pathology and monitor disease progression can be grouped into 3 categories: aggregate Aβ, aggregated tau, and neurodegeneration. It is important to note that although abnormal biomarkers of aggregate Aβ and tau are used to define AD neuropathology, the sequencing of these abnormalities and the role they play in causing cognitive and functional decline is still under investigation. Biomarkers of neurodegeneration are general indicators of neuronal injury and are not specific to AD.21
Consistent with the amyloid cascade hypothesis, it is generally believed that aggregate Aβ biomarkers represent the earliest signs of AD-related neuropathologic changes. Although changes in Aβ aggregation alone are not considered enough to cause cognitive decline, they may be sufficient to cause downstream accumulation of tau, neurodegeneration, and ultimately cognitive decline (TABLE 2).21
| Table 2. Overview of Biomarkers Used to Measure AD-Related Neuropathological Changes21 |
Biomarker |
Biomarker Category |
Aggregate Aβ
(Aβ pathology) |
Aggregated tau
(NFT)
(tau pathology) |
Neurodegeneration
(neurotoxicity) |
| CSF Aβ42 |
X |
|
|
| Amyloid PET |
X |
|
|
| CSF p-tau |
|
X |
|
| Tau PET |
|
X |
|
| Anatomic MRI |
|
|
X |
| FDG PET |
|
|
X |
| CSF t-tau |
|
|
X |
| Abbreviations: Aβ, amyloid-beta; AD, Alzheimer’s disease; CSF, cerebrospinal fluid; FDG, fluorodeoxyglucose; MRI, magnetic resonance imaging; NRT, neurofibrillary tangle; PET, positron emission tomography; p-tau, phosphorylated tau; t-tau, total tau. |
Diagnostic Guidelines
In 2011, the National Institute on Aging and the Alzheimer's Association (NIA-AA) proposed separate sets of diagnostic guidelines for symptomatic stages of AD (MCI and dementia) and a new stage for AD patients without symptoms called “preclinical AD.” The recommendations for preclinical AD were designed to aid AD research to facilitate the identification and staging of study participants who had abnormal AD biomarkers but were not cognitively impaired (FIGURE 2). 13
| Figure 2. NIA-AA Proposed Preclinical Alzheimer’s Disease Staging Framework13 |
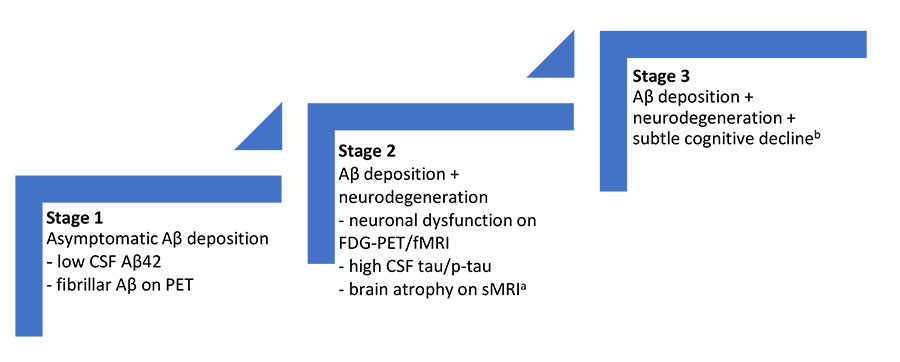 |
Abbreviations: AD, Alzheimer's disease; Aβ, amyloid-beta; CSF, cerebrospinal fluid; FDG, fluorodeoxyglucose, fMRI, functional magnetic resonance imaging; MCI, mild cognitive impairment; NIA-AA, National Institute on Aging and the Alzheimer’s Association; PET, position emission tomography; p-tau, phosphorylated tau; sMRI, structural magnetic resonance imaging.
aCortical thinning/hippocampal atrophy on sMRI.
bEvidence of subtle change from baseline level of cognition/poor performance on more challenging cognitive tests/does not yet meet criteria for MCI. |
The 2011 NIA-AA guidelines were updated in 2018 to incorporate new biomarker findings and reinforce earlier findings that certain imaging and cerebrospinal fluid (CSF) biomarkers are valid markers of AD-related neuropathological changes. Studies comparing imaging-to-autopsy results showed that Aβ positron emission tomography (PET) is a valid surrogate marker of Aβ deposition in the brain, and CSF Aβ42 is now accepted as a valid marker of abnormal Aβ pathology. Positron emission tomography ligands for tau pathology were also introduced. These updated guidelines also highlight the fact that biomarkers of neurodegeneration commonly used in AD research such as magnetic resonance imaging (MRI), fluorodeoxyglucose (FDG) PET, and CSF total tau (t-tau) are not specific for AD but may instead be indicators of neuronal damage from other causes such as cerebrovascular injury.21
Aducanumab
Aducanumab is a human IgG1 anti-Aβ monoclonal antibody directed against aggregated soluble (oligomer) and insoluble (fibril and plaque) forms of Aβ. Before its approval in June 2021, there were no FDA-approved, disease-modifying treatments in the US and no AD treatments approved since 2003.3 Several reasons have been proposed to explain the failure of previously studied AD disease-modifying therapies, including the initiation of therapies too late in the disease progression, inappropriate dosing, wrong primary therapy target, and a lack of understanding of the pathophysiology of AD.22
The research pipeline for AD-focused treatments is encouraging. An annual review of the AD drug development pipeline conducted in 2021 identified 126 therapies in 152 trials evaluating new therapies for AD. Most medications in these trials (82.5%) were disease-modifying therapies, 10.3% focused on enhancing cognition, and 7.1% were being developed to reduce neuropsychiatric symptoms.23 Disease-modifying therapies generally target either Aβ pathology (immunotherapies and inhibitors of γ-secretase and β-secretase enzymes) or tau pathology (tau aggregation inhibitors, tau phosphorylation inhibitors, and tau immunotherapy) (FIGURE 3).2
The FDA accelerated approval pathway is a mechanism by which investigational treatments that fill an unmet medical need, such as AD, can be approved based on a surrogate endpoint (biomarker) that is thought to predict clinical benefit.24 Aducanumab was approved using this pathway, and the biomarker used to support its approval was Aβ plaque reduction in the brain.3,25
Aducanumab's approval was based on findings from 3 studies: a phase 1 PRIME trial (N = 197) (NCT01677572) and two phase 3 double-blind, randomized, placebo-controlled trials: EMERGE (Study 1, NCT02484547) and ENGAGE (Study 2, NCT02477800) (N = 3285).25 To be included in the phase 3 trials, patients were required to have a positive PET Aβ brain scan and either MCI due to AD (>80% of trial participants) or mild AD. In addition, patients must have had a baseline Mini-Mental State Examination (MMSE) score of 24-30 to enroll in the phase 3 trials. The primary endpoint used in these trials was a measure of cognitive decline captured using the Clinical Dementia Rating–Sum of Boxes (CDR-SB).3,25 The CDR-SB includes scores from 6 domains: 3 domains of cognition (memory, orientation, judgment/problem-solving) and 3 domains of function (community affairs, home/hobbies, personal care). The “sum of boxes” score is the total from the 6 domains with values ranging from 0 to 18. Higher scores indicate greater disease severity.10
PET imaging was used as a biomarker to evaluate the effect of aducanumab on Aβ pathology. PET imaging compared Aβ plaque levels in regions of the brain that are expected to be widely affected by AD pathology to areas that are expected to be spared from the disease. Biomarkers used to evaluate the treatment effect of aducanumab on tau pathology were CSF p-tau and tau PET. CSF t-tau was used as a biomarker to assess the impact of treatment on downstream neurodegeneration.25
At baseline, the age ranges of trial participants in Study 1 and Study 2 were 50 to 85 years, with a mean age of 71 years.25 Study 1 and Study 2 had conflicting results, and both studies were terminated early due to their inability to achieve study objectives. At week 78, Study 1 met the primary endpoint (change from baseline on the CDR-SB) in the high dose (10 mg/kg) group with a placebo-adjusted difference of -0.39 (-22%, P = .01). However, this change was not significantly different in the low dose (3 or 6 mg/kg) group with a placebo-adjusted difference of -0.26 (-15%, P = .09). Study 2 failed to detect a statistically significant difference from placebo in either the high-dose (P = .83) or low-dose (P = .23) groups for this clinical endpoint.3,25 In all 3 trials, treatment with aducanumab resulted in a dose- and time-dependent reduction in Aβ plaques (TABLE 3). In Study 1 and Study 2, treatment reduced biomarkers of tau pathology and neurodegeneration (TABLE 4).25 Based on these findings, the recommended maintenance dose of aducanumab is 10 mg/kg given as an intravenous (IV) infusion every 4 weeks.
| Table 3. High-Dose Aducanumab Aβ PET Results (Study 1, Study 2, PRIME Study)25 |
Amyloid Biomarker
at Week 78a |
Aducanumab
(High dose, 10 mg/kg) |
Placebo |
| EMERGE (Study 1) |
| Aβ PET Composite SUVR |
N = 170 |
N = 159 |
| Mean baseline |
1.383 |
1.375 |
| Change from baseline |
-0.264 |
0.014 |
| Difference from placebo |
-0.278, P < .0001 |
| Aβ PET Centiloid |
N = 170 |
N = 159 |
| Mean baseline |
85.3 |
83.5 |
| Change from baseline (%) |
-60.8 (-71%) |
3.4 |
| Difference from placebo |
-64.2, P < .0001 |
| ENGAGE (Study 2) |
| Aβ PET Composite SUVR |
N = 183 |
N = 204 |
| Mean baseline |
1.407 |
1.376 |
| Change from baseline |
-0.235 |
-0.003 |
| Difference from placebo |
-0.232, P < .0001 |
| Aβ PET Centiloid |
N = 183 |
N = 204 |
| Mean baseline |
90.8 |
83.8 |
| Change from baseline (%) |
-54.0 (-59%) |
-0.5 |
| Difference from placebo |
-53.5, P < .0001 |
| PRIME Study |
| Aβ PET Composite SUVR |
N = 28 |
N = 42 |
| Mean baseline |
1.432 |
1.441 |
| Change from baseline |
-0.263 |
0.014 |
| Difference from placebo |
-0.277, P < .0001 |
| Aβ PET Centiloid |
N = 28 |
N = 42 |
| Mean baseline |
94.5 |
96.5 |
| Change from baseline (%) |
-58.0 (-61%) |
3.1 |
| Difference from placebo |
-61.1, P < .0001 |
Abbreviations: Aβ, amyloid-beta; PET, positron emission tomography; SUVR, standardized uptake value ratio.
aP-values were not statistically controlled for multiple comparisons. |
| Table 4. High-Dose Aducanumab Tau Biomarker Results (Study 1 and Study 2)25 |
Tau Biomarker
at Week 78 |
Aducanumab
(High dose, 10 mg/kg) |
Placebo |
| EMERGE (Study 1) |
| CSF p-taua (pg/mL) |
N = 17 |
N = 28 |
| Mean baseline |
100.11 |
72.55 |
| Change from baseline |
-22.93 |
-0.49 |
| Difference from placebo |
-22.44, P = .0005 |
| CSF t-taub (pg/mL) |
N = 17 |
N = 28 |
| Mean baseline |
686.65 |
484.00 |
| Change from baseline |
-112.44 |
-0.39 |
| Difference from placebo |
-112.05, P =0.0088 |
| ENGAGE (Study 2) |
| CSF p-taub (pg/mL) |
N = 18 |
N=15 |
| Mean baseline |
121.81 |
94.53 |
| Change from baseline |
-13.19 |
-2.24 |
| Difference from placebo |
-10.95, P = .30 |
| CSF t-taub (pg/mL) |
N = 16 |
N = 14 |
| Mean baseline |
618.50 |
592.57 |
| Change from baseline |
-102.51 |
-33.26 |
| Difference from placebo |
-69.25, P = .31 |
Abbreviations: CSF, cerebrospinal fluid; pg/mL, picograms per milliliter; p-tau, phosphorylated tau; t-tau, total tau. Note: P-values were not statistically controlled for multiple comparisons.
aBiomarker of tau pathology.
bBiomarker of downstream neurodegeneration. |
Amyloid-Related Imaging Abnormalities
Clinical trials of some but not all anti-Aβ monoclonal antibodies, particularly those targeting aggregated and or deposited Aβ, have found that these therapies can cause amyloid-related imaging abnormalities (ARIA). The biological mechanism of ARIA is not well understood. ARIA-edema (ARIA-E) can be observed on MRI as brain edema and ARIA-hemosiderin deposition (ARIA-H), includes microhemorrhage and superficial siderosis.10 It is also important to note that concurrent use of medications with anticoagulant or antiplatelet properties (other than low-dose aspirin) were not allowed in aducanumab’s clinical trials which could affect generalizability of the results to all patients.
ARIA generally occurs early during treatment, is asymptomatic, and is more common in APoE4 carriers.10 ARIA was a safety concern raised during aducanumab's FDA review process and is listed as a warning in the package insert.10,25 Among the 1105 patients treated with high-dose aducanumab during Study 1 and Study 2, 454 (41%) experienced ARIA (-E and/or -H), compared to 10% of placebo-treated patients.25 Approximately one-quarter of all patients who experienced ARIA reported associated symptoms including headache, confusion, dizziness, and nausea.25,26 Serious symptoms associated with ARIA were reported in 0.3% of patients treated with high-dose aducanumab.25 Other common adverse effects reported in aducanumab-treated patients during the clinical trials were headache (21%), fall (15%), diarrhea (9%), and confusion/delirium/altered mental status/disorientation (1%).25
Aducanumab approval has been controversial. Some experts question if reducing Aβ plaques in AD patients to levels seen in scans of healthy individuals results in a clinically meaningful slowing of cognitive decline.27 As part of the accelerated approval process, aducanumab's manufacturer must conduct a postapproval trial to verify the clinical benefits of treatment. These results will help address the question about clinically meaningful benefits.28 Three other anti-Aβ monoclonal antibodies currently in late-stage clinical development have shown promising results on reducing Aβ plaques and were granted accelerated approval status in 2021. These include donanemab, lecanemab (BAN2401), and gantenerumab. All 3 monoclonal antibodies target aggregate forms of Aβ.2,10
Emerging Therapies
Aβ peptides are produced after the sequential cleavage of the large precursor protein APP by the enzymes γ-secretase and β-secretase.16,22 Inhibition of these enzymes has been studied as a potential mechanism to reduce Aβ peptide production.2 The γ-secretase enzyme is not specific to APP. Instead, it acts on other proteins, which play an essential role in controlling normal cell growth and communication.22 Phase 2 studies of the γ-secretase inhibitors, tarenflurbil and semagacestat, initially showed promising results on reducing cognitive decline and lowering Aβ40 levels. However, in phase 3 studies, poor efficacy with tarenflurbil and safety concerns with semagacestat resulted in early study termination.2
The β-secretase enzyme acts as the rate-limiting step in Aβ production. Several small molecules that inhibit this enzyme have been studied, including lanabecestat, verubecestat, atabecestat, umibecestat, and elenbecestat. Although β-secretase inhibitors have achieved up to 80% to 90% reductions in CSF Aβ in patients with mild-to-moderate AD in clinical trials, they have failed to demonstrate significant slowing of cognitive decline. In addition, significant mental and clinical worsening in subjects treated with β-secretase inhibitors has resulted in early trial termination. Currently, there are no β-secretase inhibitors in late-stage clinical development.29
Several tau-targeted strategies have been studied, including tau aggregation inhibition, p-tau reduction, and tau immunotherapy. Earlier research focused on inhibiting tau aggregation and reducing p-tau but have mostly been abandoned due to toxicities and/or lack of efficacy. More recently, tau-targeting therapy research has shifted to immunotherapies.30 Several tau-targeted immunotherapies have reached phase 2 studies, including gosuranemab, tilavonemab, zagotenemab, and semorinemab. Results from preclinical trials were initially promising; however, there were only slight decreases in p-tau in human trials and no signs of cognitive improvement.2 A phase 2 clinical trial of AADvac-1, an active peptide vaccine that targets tau pathology, found the vaccine to be effective at slowing neurodegeneration and significantly reduced clinical and functional decline in study participants with mild AD.31
USHERING IN THE NEW ERA OF AD MANAGEMENT
An early and accurate diagnosis of MCI is critical, as this is the phase of the disease during which treatment with disease-modifying therapies has been studied and found to be most efficacious. The following steps have been proposed to facilitate the early identification of MCI32:
- Assess the patient’s and family concerns about changes in cognition
- Screen patients for MCI and evaluate their ability to function independently
- Rule out other potential causes
- Evaluate the impact of symptoms on the patient’s memory
- Determine if biomarkers are appropriate.32
Brief validated cognitive screening questionnaires are readily available for trained health care professionals. These questionnaires can be administered in less than 10 minutes.32 Examples include the MMSE and Montreal Cognitive Assessment (MoCA). If patients test positive for MCI on 1 of the brief screening questionnaires, more comprehensive screening questionnaires are available that generally take less than 20 minutes to administer. Examples include the CDR-SB and Alzheimer’s Disease Assessment Scale–Cognitive Subscale (ADAS-Cog).32,33 The Mini-Cog is a screening tool for early dementia that is suited for both health care and community settings. It combines the Three-Word Recall and the Clock Drawing Test.34 It requires minimal training, is less influenced by low education and literacy than the MMSE, and on average takes 3 minutes to administer compared to 7 minutes for the MMSE.35,36
Many aspects of AD overlap with other types of dementia, and it is essential to rule out other potential causes such as metabolic, endocrine, and nutritional disorders (eg, thyroid disease, vitamin B12 deficiency, and heavy metal poisoning), chronic infections, brain tumors, subdural hematoma, depression, and medications.15 In the research setting, Aβ biomarkers are available to support a diagnosis of MCI and increase the certainty that AD is the underlying pathology. As patients with MCI can improve, remain stable, or decline cognitively, biomarkers can help evaluate disease progression.4
Coverage Concerns
AJ calls the pharmacy and asks to talk with the pharmacist about the new Alzheimer's medication, aducanumab. She heard about it on the news and wants to know if her insurance will pay for it (Medicare) and if she would be a good candidate to try the medication.
AD is a late-onset disease that primarily impacts Medicare beneficiaries. Determining who should receive treatment with anti-Aβ monoclonal antibodies may ultimately be decided by the Centers for Medicare & Medicaid Services (CMS), at least in the short term. The widespread use of aducanumab and future AD-directed monoclonal antibodies could have significant financial consequences for payers and patients alike. The estimated annual cost per patient for aducanumab is $56,000.37
On January 11, 2022, CMS posted a decision memorandum that proposes limiting the coverage of anti-Aβ monoclonal antibodies to CMS-approved randomized controlled trials and trials sponsored by the National Institutes of Health (NIH). These trials will be required to be conducted in a hospital-based outpatient clinic, and all patients will be required to have an Aβ PET brain scan. In addition, to be eligible to enroll in these trials, patients must have a clinical diagnosis of MCI due to AD or mild AD dementia and evidence of Aβ pathology consistent with AD.38
If the proposal is accepted, these trials will address 2 fundamental knowledge gaps:
- Does treatment with anti-Aβ monoclonal antibodies result in a significant and clinically meaningful difference in decline in cognitive function?
- What are the adverse events associated with the use of AD anti-Aβ monoclonal antibodies?
Another knowledge gap that needs additional guidance is integrating existing symptomatic treatments safely and effectively with disease-modifying agents. Patients could continue approved therapies (cholinesterase inhibitors and memantine) for AD during aducanumab clinical trials.25 This provides at least some level of reassurance that combination use is safe.
After confirming that AJ is a Medicare beneficiary, you explain that aducanumab is an expensive medication that costs $56,000 a year. There is a proposal to make treatment available through a restricted program to Medicare beneficiaries. If the proposal is accepted, AJ would have to have a clinical diagnosis of MCI due to AD and evidence of Aβ plaques on a PET brain scan to meet study enrollment criteria.
HOW CAN PHARMACISTS SUPPORT AD MANAGEMENT?
Although aducanumab's approval has been controversial and continued approval is contingent upon verification of clinical benefit in confirmatory trials, its approval does provide a glimmer of hope for the millions of patients and their families in the US living with this progressive, irreversible disease that strips them of their memory and dignity. AD research is rapidly evolving, and multiple new treatments are in late-phase clinical development. Pharmacists can serve as a source of accurate and reliable information for their patients and fellow health professionals on ongoing clinical trials and emerging new treatment options.
Pharmacists can help their patients understand and mitigate their risk of developing AD and the likelihood of the disease progressing to dementia. Pharmacists can work with patients to help them manage modifiable AD-related risk factors such as high cholesterol, uncontrolled glucose, high blood pressure, smoking, obesity, and physical inactivity, which may help prevent or delay cognitive decline.19
Typically, the first presenting symptom of MCI is memory loss followed by impairment of reasoning, judgment, behavior, and communication.39 A pharmacist may be the first health care professional to notice early symptoms of AD in their patients and can aid in the early identification of AD-related symptoms. Pharmacists can play a pivotal role in screening patients for MCI or early onset of AD by offering cognitive memory screenings as part of their regular medication reviews.39 Early identification and referral to a physician for a complete evaluation will ensure patients receive timely and appropriate interventions.
In any pharmacy setting, during medication reviews, pharmacists can review the patient's refill history to ensure they are taking their medications as prescribed and, if needed, can recommend interventions such as a pillbox, medication reminder strategies, or prepackaged medications. If patients are confused or unable to remember or tolerate their medications, the pharmacist can refer them to their primary care provider for follow-up. Pharmacists can also inquire about the use of over-the-counter (OTC) medications including anticholinergic sleep aids. Community pharmacists can work with patients who have trouble with insurance coverage for their Alzheimer's medications by helping them find prescription discount cards. Inpatient pharmacists can monitor patients' anticholinergic burden, known to contribute to cognitive problems, and intervene with the medical team when appropriate.
Currently, there is limited information on how best to integrate aducanumab into clinical practice. For example, how should patients be screened to determine treatment eligibility, how should patients be monitored during treatment, and how long should treatment be continued. Some centers are developing screening and safety monitor protocols based on the study protocols used in the ENGAGE and EMERGE trials to address these and other treatment-related questions. Pharmacists can play a pivotal role in facilitating the development of these protocols and monitoring aducanumab's safety.
Lastly, as mandated reporters, all pharmacists should look for potential abuse in the elderly population, especially those with AD. In the community, estimates of the prevalence of abuse in those with dementia range from 5.4% to 62.3%. Abuse can be physical, financial, and can also take the form of neglect, which is a significant concern for patients with AD who often have complete dependence on others.40
REFERENCES
- Hickman RA, Faustin A, Wisniewski T. Alzheimer disease and its growing epidemic: risk factors, biomarkers, and the urgent need for therapeutics. Neurol Clin. 2016;34(4):941-953. doi:10.1016/j.ncl.2016.06.09
- García-Morales V, González-Acedo A, Melguizo-Rodríguez L, et al. Current understanding of the physiopathology, diagnosis, and therapeutic approach to Alzheimer's disease. Biomedicines. 2021;9(12):1910. doi:10.3390/biomedicines9121910
- Tampi RR, Forester BP, Agronin M. Aducanumab: evidence from clinical trial data and controversies. Drugs Context. 2021;10:2021-7-3. doi:10.7573/dic.2021-7-3
- Alzheimer’s Association. 2021 Alzheimer’s disease facts and figures. Alzheimers Dement. 2021;17(3):327-406. doi:10.1002/alz.12328
- Alzheimer’s Association. 2014 Alzheimer’s disease facts and figures. Alzheimers Dement. 2014;10(2):e47-e92. doi:10.1016/j.jalz.2014.02.001
- Alzheimer’s Association. Changing the trajectory of Alzheimer’s disease: how a treatment by 2025 saves lives and dollars. Published 2015. Accessed January 21, 2022. https://www.alz.org/media/Documents/changing-the-trajectory-r.pdf
- Birks J. Cholinesterase inhibitors for Alzheimer's disease. Cochrane Database Syst Rev. 2006;(1):CD005593. doi:10.1002/14651858.CD005593
- Kuns B, Rosani A, Varghese D. Memantine. In: StatPearls [Internet]. StatPearls Publishing; 2022. Updated September 14, 2021. https://www.ncbi.nlm.nih.gov/books/NBK500025/
- Hurd MD, Martorell P, Delavande A, et al. Monetary costs of dementia in the United States. N Engl J Med. 2013;368:1326-1334. doi:10.1056/NEJMsa1204629
- Combined FDA and Applicant PCNS Drugs Advisory Committee Briefing Document. November 6, 2020. Accessed January 31, 2022. https://fda.report/media/143503/PCNS-20201106-CombinedFDABiogenBackgrounder_0.pdf
- Garre-Olmo J. Epidemiology of Alzheimer's disease and other dementias [article in Spanish]. Rev Neurol. 2018;66(11):377-386.
- What happens to the brain in Alzheimer’s disease? National Institute on Aging. U.S. Department of Health & Human Services. Accessed January 21, 2022. https://www.nia.nih.gov/health/what-happens-brain-alzheimers-disease
- Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging–Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):280-292. doi:10.1016/j.jalz.2011.03.003
- Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging–Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):270-279. doi:10.1016/j.jalz.2011.03.008
- Barrera-Ocampo A, Lopera F. Amyloid-beta immunotherapy: the hope for Alzheimer disease? Colomb Med (Cali). 2016;47(4):203-212.
- Li S, Jin M, Koeglsperger T, et al. Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci. 2011;31(18):6627-6638. doi:10.1523/JNEUROSCI.0203-11.2011
- Hane FT, Robinson M, Lee BY, et al. Recent progress in Alzheimer's disease research, part 3: diagnosis and treatment. J Alzheimers Dis. 2017;57(3):645-665. doi:10.3233/JAD-160907
- Hanseeuw BJ, Betensky RA, Jacobs HIL, et al. Association of amyloid and tau with cognition in preclinical Alzheimer disease: a longitudinal study [published correction appears in JAMA Neurol. 2019;76(8):986]. JAMA Neurol. 2019;76(8):915-924. doi:10.1001/jamaneurol.2019.1424
- Risk factors and risk reduction. What are the risk factors for dementia and how can the risk be reduced? Alzheimer’s Disease International. Accessed January 21, 2022. https://www.alzint.org/about/risk-factors-risk-reduction/
- Livingston G, Huntley J, Sommerlad A. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet. 2020;396(10248):413-446. doi:10.1016/S0140-6736(20)30367-6
- Jack CR Jr, Bennett DA, Blennow K, et al. NIA-AA research framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14(4):535-562. doi:10.1016/j.jalz.2018.02.018
- Yiannopoulou KG, Papageorgiou SG. Current and future treatments in Alzheimer disease: An update. J Cent Nerv Syst Dis. 2020;12:1179573520907397. doi:10.1177/1179573520907397
- Cummings J, Lee G, Zhong K, et al. Alzheimer's disease drug development pipeline: 2021. Alzheimers Dement (N Y). 2021;7(1):e12179. doi:10.1002/trc2.12179
- Accelerated Approval Program. U.S. Food and Drug Administration. Accessed January 21, 2022. https://www.fda.gov/drugs/information-health-care-professionals-drugs/accelerated-approval-program
- Aduhelm (aducanumab-avwa) injection. Prescribing information. Biogen Inc; 2021.
- Salloway S, Chalkias S, Barkhof F, et al. Amyloid-related imaging abnormalities in 2 phase 3 studies evaluating aducanumab in patients with early Alzheimer disease. JAMA Neurol. 2022;79(1):13-21. doi:10.1001/jamaneurol.2021.4161
- George J. Alzheimer’s drugs race to FDA. MedPage Today. Published January 1, 2022. Accessed January 21, 2022. https://www.medpagetoday.com/neurology/alzheimersdisease/96462
- Aducanumab (marketed as Aduhelm) Information. FDA. Accessed January 21, 2022. https://www.fda.gov/drugs/postmarket-drug-safety-information-patients-and-providers/aducanumab-marketed-aduhelm-information
- Imbimbo BP, Watling M. Investigational BACE inhibitors for the treatment of Alzheimer’s disease.Expert Opin Investig Drugs. 2019;28(11):967-975. doi:10.1080/13543784.2019.1683160
- Congdon EE, Sigurdsson EM. Tau-targeting therapies for Alzheimer disease. Nat Rev Neurol. 2018;14(7):399-415. doi:10.1038/s41582-018-0013-z
- Inacio P. Alzheimer’s vaccine found safe, effective in patients with mild forms of the disease, phase 2 study finds. Alzheimer’s News Today. Published September 20, 2019. Accessed January 21, 2022. https://alzheimersnewstoday.com/2019/09/20/alzheimers-vaccine-safe-effective-mild-forms-of-disease-phase-2-study/
- Importance of early detection of Alzheimer’s disease. Identify Alzheimer’s Disease Earlier. Accessed January 21, 2022. https://www.identifyalz.com/
- Sheehan B. Assessment scales in dementia. Ther Adv Neurol Disord. 2012;5(6):349-358. doi:10.1177/1756285612455733
- Rickles NM, Skelton JB, Davis J, Hopson J. Cognitive memory screening and referral program in community pharmacies in the United States. Int J Clin Pharm. 2014;36(2):360-367. doi:10.1007/s11096-013-9904-7
- Borson S, Scanlan JM, Watanabe J, et al. Simplifying detection of cognitive impairment: comparison of the Mini-Cog and Mini-Mental State Examination in a multiethnic sample. J Am Geriatr Soc. 2005;53(5):871-874. doi:10.1111/j.1532-5415.2005.53269.x
- Borson S, Scanlan J, Brush M, et al. The Mini-Cog: a cognitive 'vital signs' measure for dementia screening in multi-lingual elderly. Int J Geriatr Psychiatry. 2000;15(11):1021-1027. doi:10.1002/1099-1166(200011)15:11<1021::aid-gps234>3.0.co;2-6
- Musiek ES, Gomez-Isla, T, Holtzman DM. Aducanumab for Alzheimer disease: the amyloid hypothesis moves from bench to bedside. J Clin Invest. 2021;131(20):e154889. doi:10.1172/JCI154889
- Monoclonal antibodies directed against amyloid for the treatment of Alzheimer’s disease. Centers for Medicare & Medicaid Services. Accessed January 21, 2022. https://www.cms.gov/medicare-coverage-database/view/ncacal-decision-memo.aspx?proposed=Y&NCAId=305.
- Rickles NM, Skelton JB, Davis J, Hopson J. Cognitive memory screening and referral program in community pharmacies in the United States. Int J Clin Pharm. 2014;36(2):360-367. doi:10.1007/s11096-013-9904-7
- Dong X, Chen R, Simon M. Elder abuse and dementia: a review of the research and health policy. Health Aff (Millwood). 2014;33(4):642-649. doi:10.1377/hlthaff.2013.1261
Back to Top