
Expired activity
Please go to the PowerPak
homepage and select a course.
Updates in the Management of ALS: Managed Care and Specialty Pharmacy Perspectives
OVERVIEW OF ALS
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disorder that affects neurons of the brain and spinal cord, favoring motor neurons in particular.1 It is also commonly known as Lou Gehrig’s disease, named after the famous baseball player afflicted by the disease.2 Other progressive neurodegenerative disorders include Alzheimer’s disease, Huntington’s disease, and Parkinson’s disease. These diseases result in neuronal death that progresses over time and, currently, there are no cures.3 ALS leads to motor neuron death, which causes skeletal muscle weakness and, ultimately, respiratory muscle paralysis leading to death.
EPIDEMIOLOGY OF ALS
The epidemiology of ALS is a focus of current research worldwide. ALS is not a nationally notifiable condition in the United States (U.S.), which presents challenges to epidemiological research. The National ALS Registry was created by legislation passed in 2008 in order to gather large amounts of data on disease epidemiology. The national registry allows collection of data on incidence, prevalence, demographics, and risk factors from national healthcare administrative databases and patient self-reporting through the registry portal.4 In general, ALS can be divided into familial type (family history of ALS) and sporadic type (no known family history of ALS), which account for 10% and 90% of cases, respectively.1 In the U.S., the overall prevalence of ALS is 3.9 per 100,000 people and it increases with age.4 Therefore, about 16,000 Americans are living with ALS at any given time. White males over 60 years of age are the most commonly affected group worldwide. According to data from the national registry, male sex, smoking, military service, and age 60 to 69 years are associated with an increased prevalence of ALS in the U.S.4,5
PATHOPHYSIOLOGY OF ALS
The cause of motor neuron death that occurs to cause ALS is unknown. Just as in other neurodegenerative diseases, it is exceedingly difficult to develop effective therapies or cures when the true causes of neuron death have not been elucidated. Genetic mutation and environmental factors, or a combination of both, are the most commonly accepted cause of ALS at this time. Genetic mutations are associated with both sporadic and familial ALS. At least 120 genetic mutations are associated with a risk of ALS. Certain genes, such as C9ORF72 and SOD1, have been implicated in both sporadic and familial ALS and this overlap increases the complexity of defining genetic causes for ALS.6 The final common pathway is interruption of cellular processes, including RNA homeostasis, protein homeostasis, and cytoskeletal dynamics.1
The location of neuronal death in patients with ALS leads directly to symptoms they will exhibit. To understand the symptoms and diagnosis of ALS, one should appreciate the difference between upper motor neuron (UMN) and lower motor neuron (LMN) function in the central nervous system. UMNs that may be affected are located in the motor cortex and frontotemporal lobe and cause spasticity/imbalance and dementia, respectively. LMNs originate in the brainstem or spinal cord and innervate skeletal muscle; damage leads to muscular weakness, atrophy, and fasciculations. Death of neurons located in the bulbar region of the brainstem commonly results in dysarthric speech, dysphagia, and emotional lability. Death of neurons in skeletal muscles results in upper/lower extremity weakness and respiratory muscle weakness and, ultimately, paralysis.1,7
CLINICAL PRESENTATION OF ALS
Patients have varied clinical presentations according to the site of symptom onset and any 2 patients will not present with the same clinical picture. Typically, initial weakness in 1 limb occurs in 80% of patients and weakness in the bulbar area accounts for the remaining 20% of patients. Muscle weakness and atrophy will spread to other regions as the disease progresses. Patients may also notice muscle fasciculations, but these are rarely bothersome. The combination of progressive limb weakness, LMN signs, spasticity, and UMN signs makes daily activities such as ambulation, holding objects to open, and feeding oneself increasingly difficult. Patients with bulbar-onset ALS may present with slurred speech or spastic dysarthria but preserved limb function. As symptoms progress, swallowing becomes more affected and uncontrollable laughing or crying, termed pseudobulbar affect, becomes more bothersome.8
In the last few decades, it has been discovered that cognitive and behavioral changes occur in up to 50% of patients with ALS. Severe cognitive and behavioral changes, collectively termed “frontotemporal lobe dementia,” are present in up to 15% of ALS patients. Mild cognitive changes may include difficulty with information retrieval and tracking, impaired attention, and difficulty with concentration. Reduced verbal fluency, as demonstrated by listing the maximum number of words starting with a particular letter in a specific time period, may occur. Behavioral changes may include irritability, stubbornness, apathy, and disinhibition. A commonly used tool to assess cognitive and behavioral changes in ALS is the ALS Cognitive Behavioral Screen (ALS CBS).9 This tool evaluates the patient’s abilities as well as their caregiver’s observations and can be administered during clinic visits and followed over time.
DIAGNOSIS OF ALS
Heterogeneity in disease presentation and progression are some of the challenges of diagnosing and treating ALS. Currently, ALS is a diagnosis of exclusion. Diagnosing ALS takes an average of 12 months from symptom onset. Reasons for delayed diagnosis include mild or intermittent symptoms, misdiagnosis, patient’s denial of symptoms, and decreased access to healthcare, among others.10 Research is currently being performed to find a specific biomarker that can be used to aid in diagnosis, disease staging, and treatment response.11 Tools used to make an accurate ALS diagnosis are a detailed patient history of symptoms, neurological examination, electromyography (EMG), and progression of symptoms over time. An EMG is part of the diagnostic procedures and involves a needle electrode inserted into the muscle to measure electrical activity. Diagnosis early in the course of the disease when the patient has symptoms limited to 1 region of the body may be difficult and depends on progression to other regions. Muscle weakness, which is often on 1 side of the body, or changes in the voice or speech are typical initial features of the illness. The most commonly used diagnostic scheme is the El Escorial criteria, which was created for use in clinical trials.12
Diagnosis of ALS requires the presence of:
- Evidence of LMN degeneration by clinical, electrophysiological, or neuropathologic examination
Examples: muscle atrophy, muscle weakness, and fasciculations (i.e., muscle twitches)
- Evidence of UMN degeneration by clinical examination
Examples: brisk reflexes and muscle spasticity
in addition to:
3) Progressive spread of symptoms or signs within a region or to other regions, as determined by history or examination,
together with the absence of:
1) Electrophysiological or pathological evidence of other disease processes that might explain the signs of LMN and/or UMN degeneration, and
2) Neuroimaging evidence of other disease processes that might explain the observed clinical and electrophysiological signs.
Patients are then given a diagnosis of suspected, possible, probable, or definite ALS. A panel revised the El Escorial criteria in 2015 on the basis of data showing a low probability of false diagnosis in patients deemed as “possible” ALS and current diagnostic workup practices.13 These simplified criteria are:
- Progressive UMN and LMN deficits in at least 1 limb or region of the human body (i.e., meeting the revised El Escorial criteria for possible ALS).
- LMN deficits as defined by clinical examination (1 region) and/or by EMG in 2 body regions (defined as bulbar, cervical, thoracic, and lumbosacral). The EMG findings consist of neurogenic potentials and fibrillation potentials and/or sharp waves.
Other criteria such as the Awaji criteria have been studied but do not enjoy widespread use.14 Importantly, the diagnosis should be revisited frequently if the patient fails to progress as expected or develops atypical features.10
PROGRESSION OF ALS
The rate of disease progression as described by loss of function varies widely in the ALS population. The most commonly utilized tool to quantify disease progression in clinical trials is the ALS Revised Functional Rating (ALFRS-R) Scale. It is relatively quick and easy to use. Patients or caregivers can answer questions assessing the patient’s loss of muscle function in each of 12 domains (Table 1).15 The patient is assigned a score of 4 (for no loss of function) to 0 (for significant impairment) in each domain with a maximum score of 48. The higher the score, the better the daily functioning of the patient. Limitations of this tool include lack of sensitivity to detect slow progression and influence of patient’s mood and outlook.16
| Table 1: Domains Evaluated in ALFRS-R Score15 |
| Salivation |
Speech |
| Handwriting |
Swallowing |
| Cutting food |
Walking |
| Turning in bed |
Dyspnea |
| Dressing and hygiene |
Climbing stairs |
| Orthopnea |
Respiratory insufficiency |
| ALFRS-R, ALS Revised Functional Rating Scale. |
Respiratory function should be evaluated frequently. Forced vital capacity (FVC) and negative inspiratory force (NIF) are key parameters that can be used to detect respiratory insufficiency. Other respiratory parameters may be preferred according to local clinician preference.17
PROGNOSIS OF ALS
The life expectancy for a person with ALS is typically 3 to 5 years. However, according to epidemiological data, up to 20% of patients survive to 5 years or longer and up to 10% of patients survive for 10 years or longer.18
There is no generally accepted prognostic tool for ALS.19 The heterogeneity in survival has many implications, from patient counseling to clinical trial design. For example, it is becoming more common that clinical trials for ALS have a pre-trial observation period to assess baseline loss of function prior to trial interventions.20
There is no cure that completely stops or reverses the progression to respiratory failure and death. The goals of care for ALS are to slow progression of functional decline and maintain or improve the patient’s quality of life.
TREATMENT FOR ALS
There are currently 2 medications that are approved by the U.S. Food and Drug Administration (FDA) to slow the progression of ALS: riluzole and edaravone. However, pharmacologic treatment is not the only intervention shown to slow disease progression or prolong survival.
Specialized multidisciplinary care in an ALS clinic has been shown to prolong survival and improve quality of life.21 Members of the multidisciplinary team should include respiratory therapists, social workers, speech therapists, dieticians, occupational therapists, nurse case managers, and physicians. Such specialized clinics are not available in all geographic regions, but increased utilization of telehealth services could expand the number of patients with ALS able to receive this level of care.
Non-invasive positive pressure ventilation (NIPPV) is recommended to prolong survival10 and slow respiratory decline17 in patients who have signs of respiratory insufficiency. Non-compliance with NIPPV is common and should be met with education on its benefits, adjustment of equipment, and reintroduction at later visits. Patients may be more compliant if the concept and NIPPV equipment are introduced early in the disease course.17
Adequate nutrition is key to prevent weight loss in patients with ALS since malnutrition is associated with shortened survival.22 Weight loss associated with swallowing difficulty can be mitigated by adding nutritional supplements and altering food consistency and textures to avoid choking.17 However, patients should be educated early on in the benefits of enteral nutrition received via percutaneous endoscopic gastronomy (PEG) tube to supplement oral intake. PEG tube placement is best prior to significant respiratory insufficiency since it is a surgical procedure requiring anesthesia.
Riluzole
Riluzole was approved by the FDA in 1995 to treat ALS.23 The mechanism of action in ALS is unknown, but riluzole inhibits glutamate release and affects the activity of sodium, calcium, and potassium ion channels.24
Riluzole’s efficacy was demonstrated in 2 placebo-controlled trials. Both trials included patients aged 18 or 20 to 75 years with probable or definite ALS of no more than 5 years duration, FVC of at least 60% predicted, and liver enzymes less than or equal to 2 times the upper limit of normal (ULN). The first trial25 included 155 patients randomized to placebo or riluzole 100 mg daily for 12 months. The primary endpoint was tracheostomy-free survival and rates of change in functional status at 12 months. Secondary outcomes were rate of change in respiratory function, muscle strength, and patient perceptions of symptoms. No significant difference was seen in the primary outcome at 12 months due to the exclusion of 24 patients. There was a significant difference for patients followed to 18 months, with 49% alive in the riluzole group compared with 37% alive in the placebo group (p=0.046). The median survival was 83 days longer for patients receiving riluzole than for those receiving placebo (532 days vs. 449 days). Of the remaining outcomes, the only significant result was 33% slower deterioration in muscle function at 12 months (p=0.028).
The subsequent trial26 included 959 patients and had similar inclusion criteria to the first. Patients were randomized to 50 mg, 100 mg, or 200 mg daily of riluzole or placebo therapy for 18 months. The primary endpoint was tracheostomy-free survival and secondary endpoints were rates of change in respiratory function, muscle strength, and functional status. The unadjusted relative risk of tracheostomy-free survival at 18 months for patients receiving riluzole (any dose) compared with placebo was 0.95 (95% confidence interval [CI]: 0.91-0.99, p=0.04). When adjusted for predefined prognostic factors such as age and disease duration, the relative risk of tracheostomy-free survival at 18 months was 0.65 (95% CI: 0.5-0.85, p=0.002) for the 100-mg daily dose. The difference with the 200-mg daily dose was also statistically significant, but this dose was associated with more adverse effects, such as asthenia, dizziness, gastrointestinal issues, and liver abnormalities. There were no significant differences in secondary outcomes.
Since its approval, multiple analyses have shown benefit in patients receiving riluzole.27,28 Therefore, national guidelines recommend clinicians offer riluzole to patients diagnosed with ALS to slow disease progression.17
The recommended dosing is one 50-mg tablet by mouth twice daily.23 It is available in brand and generic oral tablets and brand liquid. Pharmacist counseling points should include avoiding administration with a high-fat meal, as it reduces absorption. The tablets can be crushed, dissolved in water, and administered via a feeding tube relatively easily.
Riluzole safety
Riluzole is generally well tolerated. The most common side effects reported clinically by patients are dizziness, nausea, and circumoral paraesthesia. In the previously described trials, approximately half of patients treated with riluzole developed at least 1 alanine aminotransferase (ALT) level above normal.29 Enzyme elevations greater than 3 to 5 times ULN occurred in less than 10% of patients. The manufacturer recommends monitoring for signs and symptoms of liver dysfunction for the first 3 months of riluzole treatment and periodically thereafter with discontinuation of therapy if ALT is greater than 5 times ULN or jaundice occurs. Very rarely, neutropenia or interstitial lung disease were reported, so patients reporting febrile illness or dry cough or dyspnea may need further workup.
Edaravone
Edaravone was first approved to treat ALS in Japan in 2015.30 The FDA approved edaravone in 2017 to treat ALS on the basis of data from trials performed in Japan.31 Its mechanism as an antioxidant is the most likely mechanism in ALS.
The patient population that showed benefit from edaravone was revealed by an initial negative trial.32 The investigators then performed a randomized controlled trial with this specific subset of patients who were more likely to show benefit.20 These specific inclusion criteria are noted in bold below:
- 20 to 75 years old
- Independent living status per Japan ALS Severity Classification
- Decrease in the ALSFRS-R score of 1 to 4 during a 12-week observation period
- Definite or probable ALS according to the El Escorial criteria
- Scores of at least 2 on all 12 items of the ALSFRS-R
- FVC of at least 80%
- Duration of disease from the first symptom (any ALS symptom) of 2 years or less
Patients were randomized to placebo or edaravone for 6 cycles, which is 6 months of therapy. The dosing scheme for edaravone intravenous (IV) infusions is as follows:
Cycle 1: 60 mg edaravone IV daily for 14 days, followed by 14 days drug free
Cycles 2 to 6: 60 mg edaravone IV administered 10 of 14 days, followed by 14 days drug free
The primary endpoint was change in ALSFRS-R score from baseline to the end of the 6th cycle. Secondary endpoints included change in FVC, measures of function and muscle strength, quality of life, and a combined endpoint of time to death or disease progression. The trial included 183 patients and the change in ALSFRS-R score (defined as least-squares mean difference) was –5.01 in the edaravone group and –7.50 in the placebo group (p=0.0013). This translated to 33% less worsening of function over 6 months. For secondary endpoints, only measures of limb/bulbar function and quality of life were significantly preserved in patients receiving edaravone (p=0.039 and p=0.031, respectively).
A placebo-controlled extension study of edaravone in this ALS subset population was performed.33 Treatment was provided for 6 additional cycles (cycles 7 to 12). Patients who received edaravone in the original trial were randomized to receive edaravone or placebo and all patients who received placebo in the original trial received edaravone in this trial. In an additional 12-week open-label period (cycles 13 to 15), all patients could receive edaravone. Endpoints included change in ALSFRS-R score, FVC, measures of function and muscle strength, quality of life, and a combined endpoint of time to death or disease progression. For the 180 patients analyzed, there were no significant differences in mean change of ALSFRS-R scores, FVC, measures of function or muscle strength, or combined endpoint of time to death or disease progression.
Luo et al34 conducted a meta-analysis of data from the 3 randomized, double-blind, placebo-controlled trials available in April 2018. The mean difference between treatment and placebo groups in ALSFRS-R score during the 6-month study period was 1.63 (p=0.02) favoring edaravone. No differences in adverse effects were found. Postmarketing results from other study groups are now available since edaravone has been available for a few years.
Abraham et al35 performed a retrospective cohort study of 93 patients (22 received edaravone, 71 did not elect to receive edaravone) who did not specifically have to fit the inclusion criteria used in the edaravone trials and followed them for 7 months. The authors chose to study all patients since it is estimated that merely 7% of European patients with ALS would meet inclusion criteria used in the clinical trials.36 There was no difference in the rate of ALSFRS-R decline, muscle strength, %FVC, or rate of death. Although this study more accurately reflects clinical practice, limitations of this study include its retrospective nature, small size, and short study period.
Park et al37 performed an observational study to investigate the effects of edaravone on 22 patients with ALS located in Korea. Only 9% of patients met all inclusion criteria used in the clinical trials and 68% were also taking riluzole. Sixteen patients completed 6 cycles of edaravone. The mean decline in ALFSR-R was 5.75 and the average change in FVC was –8.7%. No serious adverse effects led to therapy discontinuation. Skin rash occurred in 2 patients and was managed by pre-treatment with antihistamine and corticosteroids; transient leukopenia occurred in 1 patient. This study is limited by its observational nature, small size, and short study period.
Jackson et al38 surveyed providers caring for ALS patients in the U.S. regarding their experiences during the first year of edaravone availability. Most patients received edaravone infusions at home and the majority received it via port access. The average duration of therapy was 6.5 months and the majority of patients (67%) were taking concomitant riluzole. Reported adverse effects were similar to the clinical trials except for 1 report of anaphylaxis, 13 reports of thromboembolism, and 9 reports of injection site infection. Since this information is subject to reporting bias and there was no control group for comparison, these adverse events should be interpreted with caution.
There are several important things to note regarding how these trials were conducted. The requirement for an ALSFRS-R change of 1 to 4 points in a 12-week pre-observation period is becoming more common.20 As discussed previously, ALS progression is not linear nor predictable in all patients, so applying this inclusion criterion may be challenging in clinical practice. Unlike riluzole trials, these trials were neither designed nor powered to detect differences in survival as a single endpoint during the study period. Instead, the combined endpoint of death or disease progression was used. No difference in this endpoint occurred in the trials and this could partially be due to the length of the study periods of 6 months20 and 12 months.33 Lastly, it is important to note that the length of the double-blind placebo-controlled period when a group of patients were never exposed to edaravone was 6 months. The extension study had a placebo group, but these patients had received edaravone in the initial 6-cycle study period. The trials did not include patients who only received placebo for all cycles in both studies. The investigators attribute this study design to the ethical issue of patients with known ALS receiving placebo.33
Edaravone safety
Overall, edaravone is well tolerated. The most common adverse effects according to the edaravone package insert were contusion, gait disturbance, and headache.31 Caution should be used in patients with asthma, since the edaravone solution for IV administration contains sodium bisulfite, but this is not a contraindication. The most common adverse effects reported by patients receiving edaravone in the extension study33 were constipation, dysphagia, nasopharyngitis, and gait disturbance. Other post-marketing adverse effects mentioned earlier include rash, anaphylaxis, thromboembolism, and injection site infection.
THE ROLE OF SPECIALTY AND MANAGED CARE PHARMACISTS
Both riluzole and edaravone are FDA approved to treat patients with a diagnosis of ALS. Once the diagnosis is made, patients should have the opportunity to discuss with their healthcare team the appropriateness of both medications in their particular situation, and this approach is widely supported by neurology physicians.39 Since there is disparity between the broad FDA approval for edaravone in all ALS patients and the limited inclusion criteria in trials, healthcare teams and payers need to navigate the gap carefully and compassionately. Discussing trial inclusion criteria and outcomes and individual patient/family goals for treatment are vital to using these medications effectively and appropriately. It is important to note that in the trials and clinical setting, the majority of patients who received edaravone were also taking riluzole. Multiple small studies indicate the majority of patients often receive edaravone for 6 to 12 months.38 Patients may “self-select” by opting out of edaravone therapy due to personal beliefs on modifying the disease, perceived cost, or time burden. They often discontinue therapy for personal reasons such as lack of perceived benefit, progression of disease, or financial or caregiver burden.38 Aside from safety concerns, there are no data to guide discontinuation of therapy. Therefore, it is of primary importance that both riluzole and edaravone are available for patients who seek these therapies. This may be balanced with cost-management techniques such as well-considered formulary and re-authorization criteria. However, these criteria should be clear and available to the patient at the initiation of therapy to avoid unexpected therapy discontinuation and the adverse psychological consequences this can cause.
Vaccines
Guidelines recommend the seasonal influenza vaccine and pneumococcal polysaccharide vaccine 23- valent for patients with ALS.17 Since these guidelines were published prior to availability of pneumococcal conjugate vaccine 13-valent, it is reasonable to recommend this vaccine for this patient population, as well.
Optimization of medication therapy
Pharmacists can tailor medication therapy for individuals with ALS to make important improvements in their quality of life. Categories and examples for consideration are provided in Figure 1.
Figure 1: Potential Pharmacist Recommendations
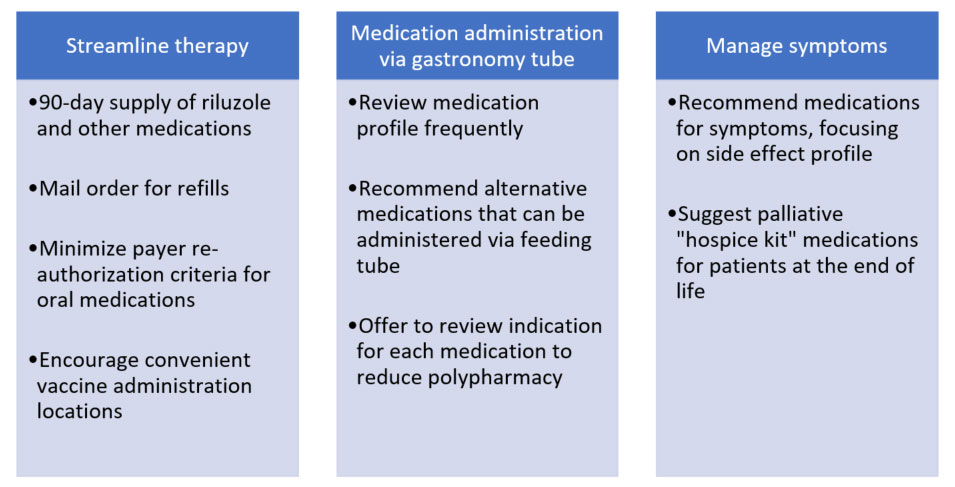
Patients with ALS are often taking medications that should not be administered via gastronomy tube. Some examples from clinical practice include long-acting formulations, capsules with pellets, and potassium chloride wax matrix tablets. Pharmacists are ideally positioned to review their medication profile with this in mind and recommend alternatives using published references.40 Early intervention to switch to alternative medications decreases stress and increases convenience for patients and their families.
Guidelines recommend treatment should be trialed for management of sialorrhea, pseudobulbar affect, cramps, spasticity, depression, anxiety, insomnia, constipation, fatigue, and pain.21 There is minimal high-quality evidence for specific medications in this setting. Therefore, pharmacists are poised to capitalize on their knowledge of dosing, side effects, and cost to recommend appropriate therapy. For example, low-dose amitriptyline (25 to 75 mg per day) can be used simultaneously to manage pseudobulbar affect, sialorrhea, and sleep disturbances due to its receptor and side effect profile. Another example is using mirtazapine for insomnia in patients who have trouble maintaining their weight since it often increases appetite.
Patients with ALS can utilize hospice services similar to patients with other terminal diseases, especially to help with in-home care.41 However, enrollment in hospice services may prohibit obtaining certain services such as power wheelchairs and may capitate medication expenses necessitating discontinuation of costly medications such as edaravone. Medications such as morphine and lorazepam prescribed and provided by their ALS care team can still benefit patients with ALS, even if they are not enrolled in a hospice program. Pharmacists can recommend appropriate selection of opiate and benzodiazepine agents and dosing while also providing vital counseling to caregivers on when and how to use these medications.
POTENTIAL FUTURE THERAPIES
Therapies for sporadic and familial ALS are currently under investigation, and stem cell therapy is a particular focus of current research.42 The 2 most common mutations associated with familial ALS – SOD1 and C9ORF72 – are the focus of current gene therapy and stem cell therapy trials.43-46 For sporadic ALS, 1 single disease target has not been identified. Gathering large amounts of data on patients who develop sporadic ALS is key. For this reason, creation of the National ALS Registry was signed into law in 2008.47 There are 2 parts to the registry: a patient portal with questionnaires regarding personal history and a tissue repository for patient samples allowing researchers to learn more about all aspects of the disease. Several studies have resulted from information provided by this database.48 Patients should be encouraged to learn more about the National ALS Registry and consider providing their information for current and future research to improve therapy or find a cure for this disease.
REFERENCES
- Brown R, Al-Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. 2017;377(12):162–72.
- Lou Gehrig and the history of ALS. ALS Association. https://www.als.org/understanding-als/lou-gehrig. Accessed June 17, 2020.
- Neurodegenerative diseases. National Institute of Environmental Health Services. https://www.niehs.nih.gov/research/supported/health/neurodegenerative/index.cfm. Reviewed September 10, 2019. Accessed June 17, 2020.
- Mehta P, Kaye W, Bryan L, et al. Prevalence of amyotrophic lateral sclerosis — United States, 2012–2013. MMWR Surveill Summ. 2016;65(8):1–12.
- Bryan L, Kaye W, Antao V, et al. Preliminary results of National Amyotrophic Lateral Sclerosis (ALS) Registry risk factor survey data. PLoS One. 2016;11(4):e0153683.
- Taylor JP, Brown RH Jr, Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016;539(7628):197–206.
- Amyotrophic lateral sclerosis. National Organization for Rare Disorders. https://rarediseases.org/rare-diseases/amyotrophic-lateral-sclerosis/. Accessed June 24, 2020.
- Tiryaki E, Horak HA. ALS and other motor neuron diseases. Continuum (Minneap Minn). 2014;20(5 Peripheral Nervous System Disorders):1185–207.
- ALS Cognitive Behavioral Screen. https://cp.neurology.org/content/neurclinpract/suppl/2017/10/11/CPJ.0000000000000397.DC1/Appendix_E-1.pdf. Accessed July 1, 2020.
- EFNS Task Force on Diagnosis and Management of Amyotrophic Lateral Sclerosis; Andersen PM, Abrahams S, Borasio GD, et al. EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS)--revised report of an EFNS task force. Eur J Neurol. 2012;19(3):360–75.
- Verber N, Shepheard S, Sassani M, et al. Biomarkers in motor neuron disease: a state of the art review. Front Neurol. 2019;10:291.
- Brooks B, Miller R, Swash M, Munsat TL; World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(5):293–9.
- Ludolph A, Drory V, Hardiman O, et al; WFN Research Group On ALS/MND. A revision of the El Escorial criteria - 2015. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16(5-6):291–2.
- de Carvalho M, Dengler R, Eisen A, et al. Electrodiagnostic criteria for diagnosis of ALS. Clin Neurophysiol. 2008;119(3):497–503.
- The ALS Functional Rating Scale. The ALS C.A.R.E. Program. https://www.outcomes-umassmed.org/als/alsscale.aspx. Accessed June 24, 2020.
- Rutkove SB. Clinical measures of disease progression in amyotrophic lateral sclerosis. Neurotherapeutics. 2015;12(2):384–93.
- Miller RG, Jackson CE, Kasarskis EJ, et al; Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: drug, nutritional, and respiratory therapies (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009;73(15):1218–26.
- Chiò A, Logroscino G, Hardiman O, et al; Eurals Consortium. Prognostic factors in ALS: a critical review. Amyotroph Lateral Scler. 2009;10(5-6):310–23.
- Creemers H, Grupstra H, Nollet F, et al. Prognostic factors for the course of functional status of patients with ALS: a systematic review. J Neurol. 2015;262(6):1407–23.
- Writing Group; Edaravone (MCI-186) ALS 19 Study Group. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017;16(7):505–12.
- Miller RG, Jackson CE, Kasarskis EJ, et al; Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: multidisciplinary care, symptom management, and cognitive/behavioral impairment (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009;73(15):1227–33.
- Desport J, Preux P, Truong T, et al. Nutritional status is a prognostic factor for survival in ALS patients. Neurology. 1999;53(5):1059–63.
- Lexicomp database. Accessed June 2, 2020.
- Wang S-J, Wang K-Y, Wang W-C. Mechanisms underlying the riluzole inhibition of glutamate release from rat cerebral cortex nerve terminals (synaptosomes). 2004;125(1):191–201.
- Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. 1994;330(9):585–91.
- Lacomblez L, Bensimon G, Leigh PN, et al. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet. 1996;347(9013):1425–31.
- Miller R, Mitchel J, Moore D. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev. 2012;2012(3):CD001447.
- Hinchcliffe M, Smith A. Riluzole: real-world evidence supports significant extension of median survival times in patients with amyotrophic lateral sclerosis. Degener Neurol Neuromuscul Dis. 2017;7:61–70.
- Rilutek tablets [prescribing information]. Bridgewater, NJ: sanofi-aventis U.S. LLC; 2012.
- Six countries have approved edaravone for ALS treatment: NMPA approved Japan-originated treatment Edaravone in China [news release]. Osaka, Japan: Mitsubishi Tanabe Pharma; August 7, 2019. https://www.mt-pharma.co.jp/e/release/nr/2019/pdf/e_MTPC190807.pdf. Accessed July 8, 2020.
- Radicava [prescribing information]. Jersey City, NJ: Mitsubishi Tanabe Pharma Corporation; 2018.
- Abe K, Itoyama Y, Sobue G, et al; Edaravone ALS Study Group. Confirmatory double-blind, parallel-group, placebo-controlled study of efficacy and safety of edaravone (MCI-186) in amyotrophic lateral sclerosis patients. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15(7-8):610–7.
- Writing Group on behalf of the Edaravone (MCI-186) ALS 17 Study Group. Exploratory double-blind, parallel-group, placebo-controlled extension study of edaravone (MCI-186) in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(sup1):20–31.
- Luo L, Song Z, Li X, et al. Efficacy and safety of edaravone in treatment of amyotrophic lateral sclerosis-a systematic review and meta-analysis. Neurol Sci. 2019;40(2):235–41.
- Abraham A, Nefussy B, Fainmesser Y, et al. Early post-marketing experience with edaravone in an unselected group of patients with ALS. Amytroph Lateral Scler Frontotemporal Degener. 2019;20(3-4):260–3.
- Hardiman O, van den Berg LH. Edaravone: a new treatment for ALS on the horizon? Lancet Neurol. 2017;16(7):490–1.
- Park J-M, Kim S-Y, Park D, Park J-S. Effect of edaravone therapy in Korean amyotrophic lateral sclerosis (ALS) patients. Neurol Sci. 2020;41(1):119–23.
- Jackson C, Heiman-Patterson T, Kittrell P, et al. Radicava (edaravone) for amyotrophic lateral sclerosis: US experience at 1 year after launch. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20(7-8):605–10.
- ENCALS Statement on Edaravone [news release]. European Network for the Cure of ALS; July 2017. https://www.encals.eu/wp-content/uploads/2017/08/ENCALS-statement-on-edaravone-FINAL.pdf. Accessed on July 8, 2020.
- White R, Bradman V. Handbook of Drug Administration via Enteral Feeding Tubes. 3rd Pharmaceutical Press; 2015.
- FYI: hospice. ALS Association. https://www.als.org/navigating-als/resources/fyi-hospice. Published June 10, 2020. Accessed July 10, 2020.
- Identifier NCT03482050. U.S. National Library of Medicine. https://clinicaltrials.gov/show/NCT03482050. Accessed July 16, 2020.
- Hardiman O, van den Berg LH. The beginning of genomic therapies for ALS. N Engl J Med. 2020;383(2):180–1.
- Miller T, Cudkowicz M, Shaw PJ, et al. Phase 1-2 trial of antisense oligonucleotide tofersen for SOD1N Engl J Med. 2020;383(2):109–19.
- Mueller C, Berry JD, McKenna-Yasek DM, et al. SOD1 suppression with adeno-associated virus and microRNA in familial ALS. N Engl J Med. 2020;383(2):151–8.
- Identifier NCT03626012. U.S. National Library of Medicine. https://clinicaltrials.gov/show/NCT03626012. Accessed June 9, 2020.
- National Amyotrophic Lateral Sclerosis (ALS) Registry: frequently asked questions. Centers for Disease Control and Prevention. https://www.cdc.gov/als/ALSFAQ.html. Reviewed April 25, 2017. Accessed July 10, 2020.
- National Amyotrophic Lateral Sclerosis (ALS) Registry: ALS research notification for clinical trial studies. https://www.cdc.gov/als/ALSResearchNotificationClinicalTrialsStudies.html. Reviewed June 3, 2020. Accessed July 10, 2020.
Back to Top