
Expired activity
Please go to the PowerPak
homepage and select a course.
Optimizing Use of Biologic DMARDs in Rheumatoid Arthritis
INTRODUCTION
Rheumatoid arthritis (RA) is an autoimmune disorder that presents as inflammation of the synovial tissue and progressive bone erosion.1 Globally, RA affects about 0.24% of the population.2 Women are 3 times more likely than men to develop RA, with the highest prevalence of RA being in women older than 65 years.3,4 Patients with RA will commonly have pain and stiffness in multiple joints, prodromal anorexia, weakness, or fatigue.2,5 RA develops over time, with symptom onset occurring gradually over several weeks.5 Psychological implications of the disease affect many patients with RA, with up to 40% of them being diagnosed with depression. High initial disease activity, as well as female gender and comorbidities, are correlated with increased rates of depression in patients with RA.3
Due to significant disease burden, RA is associated with an increased risk of presenteeism. Assessed by using a 25-item Work Limitations Questionnaire, presenteeism is defined as workers’ presence on the job, but with reduced productivity due to disease or illness. At baseline, presenteeism in patients with RA was 3.99 hours lost per 2 work weeks, based on a work week of 28.7 hours. After 1 year, RA-related disability and deterioration in overall and mental health were significantly associated with an increased risk of presenteeism.6
While RA affects the quality of life, workplace productivity, and mental health, patients may not fully understand their disease and may not be able to properly voice their concerns to healthcare providers. Biologics have an important role in management of RA, yet many patients may not be aware of their place in therapy. Pharmacists as well as all other members of the patients’ healthcare team should actively contribute to the conversation about biologics. Assisting in agent selection, patient counseling, monitoring, and follow-up are some of the tasks that a pharmacist can perform to help increase quality of life for patients with RA.
PATHOPHYSIOLOGY AND GENERAL APPROACH TO TREATMENT
RA is an autoimmune disorder with a complex, poorly understood pathophysiology. Numerous inflammatory mediators play a role in the development of RA, most notably tumor necrosis factor alpha (TNF-alpha).7 Though slow, progress has been made in the development of agents that target a number of the underlying causes of RA.
Targeted disease modifiers, also known as biologic disease-modifying antirheumatic drugs (DMARDs), or simply as biologics, are effective but costly treatment options for patients with moderate-to-severe rheumatoid arthritis.1 With limited comparative efficacy, substantial safety concerns, and cost considerations, proper selection among the 12 available biologics (Tables 1 and 2) can prove challenging.8 Providers can choose the best agent for each individual patient by understanding the safety and efficacy profiles of these medications and other key factors such as mechanisms of action, dosing frequencies, routes of administration, and costs.
The most recent guideline recommendations from the American College of Rheumatology (ACR) come from 2015.11 Notably, two new agents have been approved since these guidelines have been published – baricitinib and sarilumab. These agents are not included in the recommendations below for that reason. In addition, because of its infrequent use in RA and a lack of further evidence beyond what was available at the time of the original 2008 ACR guideline, anakinra is not discussed in the current ACR guideline, and its use is not discussed in this monograph.9
The 2015 ACR guideline recommends the use of biologic agents in patients with early (<3 months of disease/symptoms) or established (>3 months of disease/symptoms) RA with moderate-to-high disease activity after a 3-month trial of methotrexate monotherapy or other nonbiologic DMARD therapy. Given the absence of strong data to support one biologic agent over another, the guideline simply recommends adding or switching therapy to one of the five anti-TNF agents listed in Table 1, or to a non-TNF biologic agent (the term used to collectively refer to tocilizumab, abatacept, or rituximab). In patients with established RA, tofacitinib is also an appropriate first-line biologic option. If patients do not respond to a 3-month trial of anti-TNF or non-TNF biologic therapy, the conditional recommendation is to switch therapy to a non-TNF biologic over using an anti-TNF biologic or to tofacitinib (Figure 1). Tofacitinib is recommended cautiously in the guideline simply because of a lack of long-term experience and safety data.9
| Figure 1. Treatment algorithm for rheumatoid arthritis as recommended in the 2015 ACR Guideline |
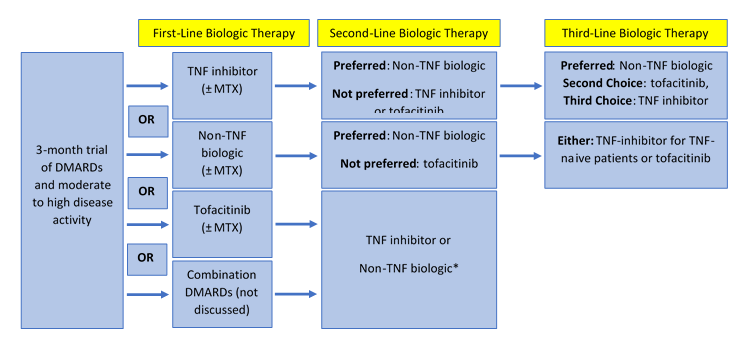 |
Information from reference 9.
*Not explicitly stated in the guideline. No recommendations are provided for patients who fail both traditional DMARD therapy and either combination DMARD therapy or tofacitinib (±MTX). |
Third-line biologic therapy should be a non-TNF biologic or tofacitinib in patients with previous anti-TNF failure, while TNF-alpha-naive patients should be treated with an anti-TNF biologic or tofacitinib. The guideline highlights that biologic therapy should be used with methotrexate, when possible, given the superior efficacy shown with the combination over biologic monotherapy.9
TNF-ALPHA INHIBITORS
While the factors that initiate the inflammatory cascade of RA are unknown, several immunomodulators are known to be involved in the damage-producing interactions that characterize the disease. Chief among these is TNF-alpha, a cytokine that mediates the detrimental interplay among activated macrophages, fibroblasts, and bone matrix. Interleukins 1 and 6 are also involved, and these provide alternative therapeutic targets for several of the non-TNF agents listed in Table 1.8
| Table 1. Available Biologic Disease-Modifying Antirheumatic Drugs |
| Mechanism of Action |
Generic Name (Trade Name) |
Route of Administration |
Dosing Frequency |
| TNF-alpha receptor antagonists |
Adalimumab (Humira) |
Subcutaneous |
40 mg every other week; 40 mg weekly can be considered for those not on concomitant methotrexate |
| |
Certolizumab pegol (Cimzia) |
Subcutaneous |
400 mg initially and at weeks 2 and 4, followed by 200 mg every other week; for maintenance dosing, 400 mg every 4 weeks can be considered |
| |
Etanercept (Enbrel) |
Subcutaneous |
50 mg once weekly with or without concomitant methotrexate |
| |
Golimumab (Simponi; Simponi Aria) |
Subcutaneous; intravenous |
Used with methotrexate. Subcutaneous: 50 mg once monthly. Intravenous: 2 mg/kg as an infusion over 30 minutes at weeks 0 and 4, then every 8 weeks |
| |
Infliximab (Remicade) |
Intravenous |
Weight-based dosing: In conjunction with methotrexate, 3 mg/kg at 0, 2, and 6 weeks, then every 8 weeks; some patients may benefit from up to 10 mg/kg or treatment as often as every 4 weeks |
| JAK enzyme inhibitor |
Tofacitinib (Xeljanz; Xeljanz XR) |
Oral |
5 mg twice daily(IR); 11 mg once daily (XR); IR should be dose reduced to 5 mg once daily with renal or hepatic dysfunction; avoid use of XR formulation in moderate-to-severe renal or hepatic impairment |
| |
Baricitinib (Olumiant) |
Oral |
2 mg once daily; use is not recommended in moderate-to-severe renal impairment or in severe hepatic impairment |
| Mediates T-cell activity by binding CD80 and CD86 |
Abatacept (Orencia) |
Subcutaneous; intravenous |
125 mg once weekly subcutaneously
Weight-based dosing for intravenous use:
Patient weight <60 kg, 500 mg (two vials)
Patient weight 60–100 kg, 750 mg (three vials)
Patient weight >100 kg, 1000 mg (four vials) |
| IL-6 receptor antagonist |
Tocilizumab (Actemra) |
Subcutaneous; intravenous |
Weight based dosing for subcutaneous administration:
• Patient weight <100 kg = 162 mg every other week; increase to weekly based on response
• Patient weight >100 kg = 162 mg every week
Intravenous doses start at 4 mg/kg as a 60-min infusion every 4 weeks with increases to 8 mg/kg every 4 weeks based on clinical response; doses can be lowered back to 4 mg/kg for dose-related adverse effects |
| |
Sarilumab (Kevzara) |
Subcutaneous |
200 mg subcutaneously once every 2 weeks |
| Binds CD20 on the surface of B lymphocytes |
Rituximab (Rituxan) |
Intravenous |
In combination with methotrexate, two 1000 mg infusions separated by 2 weeks every 24 weeks or less based on clinical evaluation, but not sooner than every 16 weeks |
| IL-1 receptor antagonist |
Anakinra (Kineret) |
Subcutaneous |
100 mg daily |
Information obtained from Reference 8.
IL - interleukin; IR – immediate-release; JAK - Janus kinase; TNF-alpha - tumor necrosis factor alpha; XR – extended-release |
| Table 2. Black-Box Warnings and Contraindications for Biologic Disease-Modifying Antirheumatic Drugs |
| Action |
Generic Name (Trade Name) |
Black-Box Warnings |
Contraindications |
| TNF-alpha receptor antagonists |
Adalimumab (Humira) |
Serious infections and malignancy |
None |
| |
Certolizumab pegol (Cimzia) |
Serious infections and malignancy |
None |
| |
Etanercept (Enbrel) |
Serious infections and malignancy |
Sepsis |
| |
Golimumab (Simponi; Simponi Aria) |
Serious infections and malignancy |
None |
| |
Infliximab (Remicade) |
Serious infections and malignancy |
Hypersensitivity to infliximab, murine proteins, or any component of the formulation; doses greater than 5 mg/kg in patients with moderate or severe heart failure (NYHA class III/IV). |
| JAK enzyme inhibitor |
Tofacitinib (Xeljanz; Xeljanz XR) |
Serious infections and malignancy |
None |
| |
Baricitinib (Olumiant) |
Serious infections and malignancy, thrombosis |
None |
| Mediates T-cell activity by binding CD80 and CD86 |
Abatacept (Orencia) |
None |
None |
| IL-6 receptor antagonist |
Tocilizumab (Actemra) |
Risk of serious infections |
Known hypersensitivity to Actemra or any component of the formulation |
| |
Sarilumab (Kevzara) |
Risk of serious infections |
Known hypersensitivity to Kevzara or any component of the formulation |
| Binds CD20 on the surface of B lymphocytes |
Rituximab (Rituxan) |
Fatal infusion reactions, severe mucocutaneous reactions, hepatitis B virus revaccination, and progressive multifocal leukoencephalopathy |
None |
| IL-1 receptor antagonist |
Anakinra (Kineret) |
None |
Known hypersensitivity to Escherichia coli-derived proteins, Kineret, or any component of the product |
Information obtained from Reference 8.
IL – interleukin; JAK – Janus kinase; NYHA – New York Heart Association; TNF-alpha – tumor necrosis factor alpha |
Introduced to the United States (U.S.) market between 1998 and 2008, five TNF-alpha inhibitors are currently available.8 Current guidelines consider these agents interchangeable. While previously thought to have similar efficacy, recent data suggest that certolizumab may be more effective than both etanercept and adalimumab when given as monotherapy or combination therapy. Certolizumab has also been shown to be more effective than golimumab and infliximab when given as combination therapy.10 However, specific TNF-alpha inhibitors may be preferred in individual patients based on routes of administration (subcutaneous versus intravenous), whether methotrexate or other nonbiologic agents are being used concomitantly, adverse effects, and patient sensitivities.
While some biologic agents are effective as monotherapy, most are more effective when combined with methotrexate.11 Of the five TNF-alpha inhibitors, infliximab and golimumab have not been shown to be effective as monotherapy in clinical trials.11-13 Given these findings, methotrexate use must be considered when using these agents. This may be problematic for some patients, as methotrexate has notable contraindications, including pregnancy and alcoholism. Methotrexate can also increase the risk of infection and can cause other dermatologic, neurologic, pulmonary, or renal complications.8 Patient appropriateness should be considered carefully since approximately one-third of patients with RA are on biologic monotherapy due to an intolerance to or nonadherence with methotrexate.14
Dosing and Administration
The routes of administration and dosing instructions of the available TNF-alpha inhibitors for RA are listed in Table 1. Infliximab is administered intravenously, golimumab is available in both subcutaneous and intravenous formulations, and the other three anti-TNF agents are administered subcutaneously. Infliximab and intravenous golimumab are dosed based on weight.8
Since many of these agents are self-administered, it is important to provide patients with manufacturer medication guides and instructions for use each time a prescription is dispensed. Patients whose joint deformity prevents self-administration or those with a history of nonadherence may benefit from intravenous administration.
General guidelines for self-administration of TNF-alpha inhibitors are shown in Table 3. Certain products may have different administration instructions compared to those listed below, so it is important to share product-specific materials with patients when dispensing any of these products.
| Table 3. Counseling Points for Subcutaneous Self-administration of TNF-alpha inhibitors |
- Store products as directed until use, generally in the refrigerator (36°–46°F [2°–8°C]).
- Remove packages from the refrigerator as directed, generally 15–30 minutes before use. Allow product to come to room temperature without warming in your hands or microwaving.
- Wash your hands with soap and water and dry them with a clean towel.
- Choose an injection site as directed, generally on the front of the thighs, the abdomen, or the upper arms.
- Sterilize the site using an alcohol pad. Allow the alcohol to dry; do not fan or blow the site.
- Remove air bubbles from syringes if directed to do so in the medication guide.
- Pinch the skin and inject the agent as directed in the medication guide.
- If bleeding occurs at the injection site, cover with a cotton ball or gauze as needed.
- Place used materials in an FDA-cleared sharps or other appropriate container and discard in accordance with state and federal laws.
|
Adverse Reactions and Tolerability
The marketed TNF-alpha inhibitors have similar adverse reaction profiles, but patients can react differently to the five agents. The most serious concerns are infection, particularly with pathogens causing tuberculosis (TB) and other potentially fatal diseases, and an increased risk of some cancers.
Infections
As immunosuppressive agents that inhibit the body's natural defenses, TNF-alpha inhibitors place patients at an increased risk of infections, including TB. However, RA itself predisposes patients to infection, complicating the assessment of risk from the drugs.15 Table 4 highlights several additional risk factors for infection in patients with RA.16
| Table 4. Risk Factors for Infection in Patients With Rheumatoid Arthritis |
- Long-term and/or high-dose corticosteroid use
- Significant comorbidity (e.g., interstitial lung disease, renal failure, diabetes mellitus)
- Surgical intervention
- Advanced age
- Connective tissue disease other than rheumatoid arthritis
- History of tuberculosis exposure
- Residing or traveling in areas with endemic diseases (e.g., tuberculosis)
- Occult hepatitis B virus
- Hepatitis C virus exposure
|
Adapted from Reference 16. |
Biologic therapy is associated strongly with an increased risk of serious infections.15,17 The true prevalence, however, may be low, with a range of 6 to 17 cases per 1000 patients per year, depending on the dose of biologic therapy.16,17 Regardless, research into this topic has shown that specific subgroups treated with biologics appear to be at a higher risk of infection than those treated with traditional DMARDs. Several points should be considered when selecting a TNF-alpha inhibitor for RA therapy:
- Methotrexate-experienced patients, those treated with combination therapy of biologics and traditional DMARDs, and those with disease duration of 2 to 10 years are at higher risk of serious infections.17
- Biologics do not increase the risk of serious infection in methotrexate-naive patients.17
- Compared with other biologics, certolizumab pegol has repeatedly shown a higher risk of serious infections,16,18,19 while etanercept has shown lower discontinuation rates and a non-significantly lower risk of hospitalization secondary to serious infection.1,19,20
Patients starting on TNF-alpha inhibitor or other biologic therapy should be monitored for signs of infection. If a patient has an active infection, the biologic treatment should be withheld until the infection is resolved.21 Once established on biologic therapy, the patient should communicate any concerns they may have to their health care providers. Since serious infections can be very problematic for those with a blunted immune response, patients must not be hesitant about opening a line of communication at the first sign of infection. Simple identifiers, such as fatigue or fever, may provide the early detection necessary to properly manage infection before it becomes severe or life-threatening. Providers should watch for elevated C-reactive protein levels and positive blood cultures with organisms of even low pathogenicity and must rule out infection immediately upon obtaining these findings.21, 22
TNF-alpha inhibitors have been associated with reactivation of latent Mycobacterium tuberculosis.15 The risk appears to be lowest among etanercept-treated patients.1,16,23 A detailed clinical history documenting potential exposure to mycobacteria and screening with interferon-γ release assays or tuberculin skin testing and a chest radiograph and acid-fast bacilli smear and culture of sputum samples in appropriate patients is necessary to ensure proper management prior to starting biologic therapy.9,16,24 If positive results are found upon screening, pretreatment with prophylactic isoniazid 300 mg/d for 9 months or combination therapy of rifapentine 900 mg and isoniazid 900 mg once weekly for 12 weeks is recommended.25,26
Patients on TNF-alpha inhibitors or other biologic therapies should be monitored for symptoms of TB including a cough lasting more than 3 weeks, hemoptysis, unexplained weight loss, fatigue, and fever. If patients present with these symptoms, the patient should be promptly evaluated by the prescriber and discontinuation of biologic therapy considered.
As with TB, hepatitis B virus (HBV) reactivation can occur in people carrying the virus who begin therapy with TNF-alpha inhibitors or other biologics for RA.16,24 HBV is a major cause of liver cirrhosis, which can lead to liver transplantation or death.16,24 A detailed clinical history is essential to identify at-risk patients, since approximately 40% of infected patients are at risk for reactivation if not treated appropriately before initiation of anti-TNF therapy.16 Product labeling for all TNF-alpha inhibitors carries a warning about HBV reactivation.8 In patients previously infected with HBV, appropriate antiviral therapy is recommended throughout treatment and for 12 months after therapy is discontinued.16
Malignancy
The risk of malignancy in patients treated with TNF-alpha inhibitors and other biologic DMARDs is controversial. The true risk of malignancy is complicated by several factors. First, there is an underlying established relationship between the immune system and cancer.27 Additionally, inflammation is a known risk factor for malignancy.24 Given that RA is an autoimmune disorder characterized by widespread inflammation, the increased risk of malignancy could be attributed to the underlying disease itself. Supporting this hypothesis, patients with RA appear to have higher rates of malignancy than the general population.24,27 However, the analysis is complicated in that patients with severe disease are the most appropriate candidates for treatment with biologic DMARDs,27 meaning that the only data available are come from patients with the highest baseline risk of malignancy.
Since TNF is predominantly a tumor-promoting cytokine,27 it stands to reason that inhibition of TNF-alpha should lower the overall cancer risk. Evidence appears to support this claim. Overall malignancy rates are not increased with 2 to 4 years of treatment with TNF-alpha inhibitors.19,24,27,28 However, patients may have concerns and should be counseled appropriately.
Patients with a history of cancer receive special attention in ACR guidelines. If it has been more than 5 years since treatment for a solid malignancy or nonmelanoma skin cancer (NMSC), any TNF-alpha inhibitor or other biologic is appropriate. Rituximab should be used in patients with a history of treated malignancy within the past 5 years.27 Other recommendations suggest no contraindication to anti-TNF therapy exists after patients have been cancer-free for 10 years.23 However, given the lack of evidence to support or refute the use of TNF-alpha inhibitors or other biologics in patients with a history of cancer, these agents must be used with caution in this population.
Anti-TNF therapy has been associated with increased risk of NMSC but not with melanoma.27,28 However, the available data are inconclusive, and the association requires significant further research.
Patients with RA have a 2- to 3-fold increased baseline risk of lymphoma.27 No further increase in lymphoma risk has been found with TNF-alpha inhibitor or other biologic DMARD therapy.16,27,28 TNF-alpha inhibitors have been shown to not increase the risk of leukemia.27 While these findings suggest the use of TNF-alpha inhibitors is not inappropriate in these patients, the ACR guideline recommends otherwise. For patients with previously treated lymphoproliferative disorders, such as lymphoma or leukemia, the guideline recommends use of rituximab over other TNF-alpha inhibitors since rituximab is indicated for some of these disorders. The guideline also mentions the use of abatacept or tocilizumab over TNF-alpha inhibitors in these patients, but does so with the stipulation that these recommendations are based on a very low level of evidence.9
TNF-alpha inhibitors also have not been shown to increase the risk of any cancers caused by oncogenic viruses, such as human papillomavirus and associated cervical and anal cancers. Anti-TNF therapy in combination with thiopurine has been linked to hepatosplenic T-cell lymphoma, but monotherapy does not share the same risk.27
Warnings and Precautions
Cardiovascular Risk
The relationship between cardiovascular complications and anti-TNF therapy is complicated by an underlying increased risk of cardiovascular events (myocardial infarction and cardiovascular-related mortality) in patients with RA.29,30 This risk is positively correlated with disease activity in that a higher level of disease activity equates to a higher risk of cardiovascular events.29 Inflammatory mediators are theorized to play a role in this increased risk. Supporting this hypothesis, anti-TNF therapy has been shown to reduce nonfatal myocardial infarction and transient ischemic attack/stroke as a composite outcome compared with nonbiologic DMARDs other than methotrexate.30
Despite a warning in product labeling for congestive heart failure (CHF) with all TNF-alpha inhibitors, the true risk is very uncertain. One large retrospective review found a decreased risk of CHF with these agents, numerous other reviews found the opposite, and others found the data inconclusive.1 The concern that these agents can cause CHF initially emerged from short-term data suggesting a high risk of new-onset or worsening heart failure with infliximab. However, data over a longer time period suggested a lower risk of cardiovascular events in patients treated with anti-TNF therapy.16 Regardless, infliximab in doses of 5 mg/kg or more is contraindicated in patients with CHF. Further, the ACR guideline suggests that TNF inhibitors should be avoided in patients with CHF and those with worsen CHF currently on a TNF inhibitor should have their therapy switched. The guideline suggests patients can be started on or switched to combination DMARDs, a non-TNF inhibitor, or tofacitinib.9
Hypertension has been shown to occur with TNF-alpha inhibitors, primarily with certolizumab pegol. Length of therapy and the risk of hypertension are positively correlated; patients on anti-TNF therapy for more than 6 months are at increased risk of hypertension.31 Blood pressure should be monitored regularly in patients during therapy with any TNF-alpha inhibitor.
Injection- and Infusion-Related Reactions
Infusion-related reactions with intravenous infliximab and injection-site reactions with the other available products are some of the most common reasons for medication discontinuation.1
Infusion reactions can be severe, anaphylactic-type reactions and occur in approximately 20% of patients treated with infliximab.16 Proper management of infusion reactions varies based on specific symptoms. For those experiencing hypertension or pruritus, decreasing the infusion rate is recommended, along with symptom control with antihypertensives or antihistamines, respectively. A slowed infusion rate also appears to be effective for patients experiencing urticaria.32
For major infusion-related reactions, including anaphylaxis, drug discontinuation is essential. Injection-site reactions tend to be less severe, but can occur with any of TNF-alpha inhibitors or other biologic agents.1 Injection-site reactions are associated with erythema, urticaria, pruritus, or rash at the injection site and occur most frequently within the first month of treatment.1,16 These reactions are very common in patients treated with etanercept, and to a lesser extent in those treated with adalimumab, certolizumab pegol, and golimumab.16 Concerned patients should be counseled that these reactions are usually self-limiting and typically do not require intervention.33 However, patients can be instructed to rotate injection sites and use antihistamines if symptoms are bothersome. The needle covers on the adalimumab, etanercept, and golimumab prefilled syringes and the etanercept and golimumab autoinjectors contain a dry natural rubber that is derived from latex. This may cause allergic reactions in sensitive individuals.
Vaccines
Vaccination coverage is surprisingly low in patients with RA, with only about 25% to 30% of patients receiving proper vaccines. This is mainly due to a lack of awareness by healthcare professionals, as well as hesitancy concerning vaccine efficacy and safety.34 The use of live vaccines is not recommended for patients with RA during treatment with any TNF-alpha inhibitor or other biologic DMARD. These medications blunt the body's ability to mount an appropriate response to live vaccines. Commonly used vaccines that should be avoided during treatment include measles, mumps, and rubella; varicella (chickenpox); zoster (herpes zoster; shingles); and live-attenuated intranasal influenza vaccine. Inactivated vaccines, such as the influenza, pneumococcal, and tetanus and diphtheria toxoid vaccines can be administered safely.16 The new herpes zoster vaccine, Shingrix, is an inactivated recombinant, adjuvanted vaccine that may be a safer option for those on TNF-alpha inhibitors compared with Zostavax, the live attenuated herpes zoster vaccine. However, concomitant administration of immunosuppressive therapies may limit the effectiveness of Shingrix.9
Key Counseling Information
As noted in previous sections, several points can be emphasized during counseling of patients on TNF-alpha inhibitors:
- Advise patients to watch closely for signs or symptoms of infection. Patients should let their prescriber or pharmacist know if any of the following symptoms occur: fevers, chills, cough lasting more than 3 weeks, coughing up blood, unexplained weight loss, fatigue, or other symptoms that could indicate the onset of infection.
- Ask about allergies to latex in patients receiving adalimumab, etanercept, or golimumab. Infusion- or injection-related skin reactions can be managed with antihistamines or, for intravenous infliximab, a slower infusion rate.
- Blood pressure should be monitored regularly and elevations reported to health providers.
- Be sure patients know to report use of these agents to all health providers and that they know not to receive live vaccines.
JAK Inhibitors
The JAK inhibitors, tofacitinib and baricitinib, are the newest novel treatments for RA. JAK inhibitors are the only orally active medications among the biologic agents. As one of the biologic agents with impressive monotherapy clinical results, tofacitinib is a valuable addition to therapy. The 2015 ACR guidelines call out tofacitinib as a preferred non-TNF biologic drug,9 but the lack of long-term safety data remains a concern. Baricitinib was approved in June 2018 and has not yet been included in the ACR guidelines. However, providers should view baricitinib and tofacitinib in the same category, as both of these agents possess similar efficacy and safety profiles.
Dosing and Administration
Tofacitinib is approved by FDA for treatment of adult patients with moderately to severely active RA who have had an inadequate response or intolerance to methotrexate as either monotherapy or in combination with methotrexate or other nonbiologic DMARDs. It is available as either an immediate-release or an extended-release product. The immediate-release product is dosed as a single 5 mg tablet twice daily, while the extended-release product is administered as a single 11 mg tablet once daily. For patients with moderate or severe renal impairment or moderate hepatic impairment, the dose of the immediate-release product is lowered to 5 mg once daily, and use of the extended-release product should be avoided.
Baricitinib is approved by FDA for treatment of adult patients with moderately to severely active RA who have had an inadequate response to one or more TNF antagonist therapies. Available as 2 mg tablets, baricitinib is administered in doses of 2 mg once daily. For patients with impaired renal function (GFR <60 mL/min/1.73 m2) or severe hepatic impairment, use of baricitinib is not recommended.
Adverse Reactions and Tolerability
The most common adverse reactions seen in patients with RA who are receiving tofacitinib are upper respiratory tract infections (4% of patients), nasopharyngitis (3%), diarrhea (3%), and headache (3%). During the first 3 months of therapy in clinical trials, 20% of patients taking 5 mg twice daily developed infections of some type, as did 18% of those in placebo groups.8
In patients with RA who are receiving baricitinib, the most common adverse reactions include upper respiratory tract infections (16% of patients), nausea (3%), increased serum ALT or AST (2% and 1%, respectively), and herpes zoster infection (1%).
Like other biologics for RA, JAK inhibitors may increase the risk of TB.1,23 The drugs should be used with caution in patients at risk for TB, but some authors suggest that pretreatment screening is not necessary unless the patient is from a TB-endemic area (see the TNF inhibitor section for details on TB prescreening and pretreatment).16 The products’ black-box warnings also list invasive fungal infections and bacterial, viral, and other infections caused by opportunistic pathogens. Use of the drug should be avoided in patients:
- With chronic or recurrent infection
- Who have been exposed to TB
- With a history of serious or opportunistic infection
- Who have resided or traveled in areas with endemic TB or mycoses
- With underlying conditions that predispose them to infections
In addition, baricitinib carries a black-box warning for serious and sometimes fatal thrombosis, including deep vein thrombosis, pulmonary embolism, and arterial thrombosis events. This agent should be avoided in patients at increased risk of thrombosis.8
Warnings and Precautions
In addition to the above concerns about serious infections, the JAK inhibitors have been associated with viral reactivations, gastrointestinal perforations, malignancies, and lymphoproliferative disorders. Patients with known malignancy other than successfully treated NMSC should not receive JAK inhibitors. Patients will need regular blood tests to monitor for low neutrophil or platelet counts or increased liver function enzymes or cholesterol.
Tofacitinib is a major substrate of cytochrome P450 (CYP450) 3A4 and a minor substrate of CYP450 2C19, while baricitinib is only a minor substrate of CYP450 3A4. Therefore, strong inducers of these pathways, primarily the CYP450 3A4 pathway, will reduce the serum concentration of tofacitinib and baricitinib to a lesser extent, while inhibitors of these pathways will increase levels. Monitor therapeutic effect and consider dose adjustment of tofacitinib in patients on strong inhibitors (e.g., clarithromycin, fluconazole, itraconazole) or strong inducers (e.g., carbamazepine, phenytoin, rifampin, St. John's wort) of these enzymes.
Key Counseling Information
Since JAK inhibitors are orally administered, patient counseling focuses primarily on adverse effects and the need for attention to potential symptoms of infections and with baricitinib, thrombotic events. Tofacitinib is contraindicated during pregnancy, and women who become pregnant while on tofacitinib should know to report their experiences in a company-sponsored registry. Baricitinib has shown harm in animal reproduction studies and therefore should be used with caution in pregnancy. Patients should also be informed of the need for blood tests to monitor effects of the drug before and during therapy and the need to avoid live vaccines.
ABATACEPT
In patients who do not respond to or tolerate TNF-alpha inhibitors, abatacept is often an appropriate next agent, generally in combination with methotrexate. Overall, some evidence indicates that its efficacy and safety profile make abatacept an optimal next choice.1,23
Acting as a selective co-stimulation modifier, abatacept binds to CD80 and CD86 on antigen-presenting cells to block their CD28 interaction with T-lymphocytes. This prevents the full activation of T-lymphocytes, thereby interrupting one of the pathways through which RA damages joints.8
Dosing and Administration
Abatacept is marketed in both subcutaneous and intravenous formulations. Intravenous infusions may be used as loading doses and regularly in those for whom adherence or self-injection is problematic.
The intravenous product is supplied in lyophilized vials containing abatacept 250 mg. The drug is reconstituted with Sterile Water for Injection 10 mL using the silicone-free disposable syringe supplied with the product and an 18- to 21-gauge needle. Intravenously, abatacept is dosed by weight: 500 mg (two vials) for patients weighing less than 60 kg, 750 mg (three vials) for patients weighing 60–100 kg, and 1000 mg (four vials) for patients weighing more than 100 kg. These amounts are infused over 30 minutes. Repeat doses are given at 2 and 4 weeks after the initial infusion and then every 4 weeks thereafter.8
When used subcutaneously, abatacept 125 mg is administered once weekly with or without an intravenous loading dose. For those transitioning from intravenous to subcutaneous administration, a subcutaneous dose should replace the next scheduled intravenous dose and be administered weekly thereafter.8 Detailed patient instructions for subcutaneous injections are provided with the product. Patients should carefully follow these instructions. The injection steps are similar to those outlined in Table 3.8
Adverse Reactions and Tolerability
As with TNF-alpha inhibitors, infections, malignancies, and infusion- or injection-related reactions are common with abatacept. Infections at slightly higher rates than with placebo were reported in clinical trials. The most common infections requiring abatacept dose interruption are upper respiratory tract infections (1% of patients), bronchitis (0.7%), and herpes zoster (0.7%). Dose discontinuation occurs most commonly with pneumonia (0.2%), localized infection (0.2%), and bronchitis (0.1%).8
Warnings and Precautions
Overall, abatacept appears to be one of the safest non-TNF biologics, with risks of serious infections such as TB no higher in those on the drug than in the general population.1,24 Other warnings in FDA-approved labeling are similar to those with other non-TNF biologics.
Patients with RA being considered for treatment with abatacept should be prescreened using the tuberculin skin test. Patients need pretreatment for TB (prophylactic isoniazid 300 mg/d for 9 months or combination therapy of rifapentine 900 mg and isoniazid 900 mg once weekly for 12 weeks) only if they are from areas where the disease is endemic16 or if prescreening indicates latent TB infection.8 Patients treated concomitantly with abatacept plus a TNF-alpha inhibitor are at increased risk of infections and serious infections such as pneumonia and septicemia; this drug combination should be avoided. During transition from TNF-alpha inhibitors to abatacept, patients should be monitored for signs of infection. Those with histories of recurrent infections or otherwise at elevated risk of infection should be treated with abatacept cautiously and new infections monitored closely. Treatment should be discontinued if serious infections occur.8
As with TNF-alpha inhibitors, hypersensitivity, anaphylaxis, and anaphylactoid reactions are a concern with abatacept therapy. During intravenous administration, treatment for severe hypersensitivity reactions should be available. If anaphylactic or other serious reactions occur, drug administration should be stopped immediately and permanently discontinued.8
As with TNF-alpha inhibitors, live vaccines should not be used during abatacept therapy and for 3 months after discontinuation. In addition, responses to inactivated vaccines may be blunted during abatacept therapy.8
The incidence of adverse events with abatacept is higher during therapy in patients with chronic obstructive pulmonary disease (COPD), with significantly higher rates than with placebo.1 Alternative biologic therapies should be considered for patients with COPD.
Key Counseling Information
As noted in previous sections, several points can be emphasized during counseling of patients on abatacept:
- Advise patients to watch closely for signs or symptoms of infection. The patient should notify the prescriber or pharmacist if fevers, chills, cough lasting more than 3 weeks, coughing up blood, unexplained weight loss, fatigue, or other symptoms that could indicate the onset of infection occur.
- Be sure patients know to report use of these agents to all health providers and that they know not to receive live vaccines.
- Patients with COPD should report worsening of symptoms, cough, and trouble breathing.
- The product should be stored in the refrigerator and removed 30 minutes before use. Patients should not attempt to accelerate the warming process in any way, including placing the product in warm water or microwaving.
IL-6 Inhibitors
Two IL-6 inhibitors are available on the market – tocilizumab and sarilumab. Both share the indication of treatment of adult patients with moderately to severely active RA after an inadequate response to prior DMARD therapy.8 The IL-6 inhibitor tocilizumab has shown much promise in clinical trials and may be the most efficacious biologic when used as monotherapy.24 Trials show the drug has been effective in patients with methotrexate-refractory RA and disease that has not responded to TNF-alpha inhibitors. Having only been approved in May 2017, sarilumab has less clinical experience to date.
Dosing and Administration
As shown in Table 1, tocilizumab is marketed in both subcutaneous and intravenous formulations. The normal intravenous dose is 4 mg/kg over 60 minutes once every 4 weeks. When transitioning from intravenous to subcutaneous administration, the first subcutaneous dose should be administered at the time when the next intravenous dose is due.8 One subcutaneous tocilizumab 162 mg injection is used for all patients, but the dosing interval varies by patient body mass: weekly for those weighing 100 kg or more, and every other week in those weighing less than 100 kg. The dosage of the intravenous product and the frequency of the subcutaneous products may be increased if patients are not receiving the expected response from therapy.8
Sarilumab is available as a 200 mg subcutaneous injection administered once every 2 weeks. Both sarilumab and subcutaneous tocilizumab should be administered in the abdomen, thighs, and upper arms. The injection sites should be rotated with each injection.8 Tocilizumab and sarilumab should be stored in the refrigerator and should be allowed to reach room temperature before administration.8
The doses of tocilizumab and sarilumab should be adjusted in patients experiencing neutropenia, thrombocytopenia, and/or acute hepatotoxicity. Guidance for specific dosage adjustments is available in the manufacturer labeling.8
Adverse Reactions and Tolerability
Adverse effects occurring in 5% or more of patients on tocilizumab monotherapy or combination therapy are upper respiratory infections, nasopharyngitis, headache, hypertension, and elevated serum liver enzymes (specifically alanine aminotransferase [ALT]). In clinical trials, similar numbers of patients discontinued therapy because of adverse effects in active drug and placebo groups. In those on active therapy, the most common reasons for discontinuation of tocilizumab were elevated liver enzymes and serious infections.
In patients taking sarilumab, the most common adverse reactions include neutropenia, increased serum ALT, injection site erythema, upper respiratory tract infections, and urinary tract infections.
IL-6 inhibitor therapy has been associated with an increased risk of TB.1,23 The drug should be used with caution in patients at risk for TB, but some authors suggest that pretreatment screening is not necessary unless the patient is from a TB-endemic area (see the TNF section for details on prescreening and pretreatment).16
IL-6 inhibitors are associated with increased cholesterol levels, including total cholesterol, low-density lipoprotein cholesterol, high-density lipoprotein cholesterol, and triglycerides.16,23 This temporary increase in lipid parameters typically resolves within 3 months of treatment initiation, though lipid-lowering therapy may be necessary in some patients. Interestingly, cardiovascular outcomes may theoretically be improved in patients receiving tocilizumab or sarilumab, since IL-6 (the therapeutic target) is thought to be involved in the development of coronary heart disease.16 Hypersensitivity reactions have occurred with IL-6 inhibitors. Patients should report any relevant symptoms and may require treatment interruptions or discontinuations.
Warnings and Precautions
Because of the adverse effects of IL-6 inhibitors, the drug should be used carefully and patients should be monitored closely for several signs and symptoms. Like most other biologic agents for RA, IL-6 inhibitors carry a black-box warning about infections. Treatment with IL-6 inhibitors increases the risk of serious infections, including TB, invasive fungal infections such as candidiasis and aspergillosis, and other infections caused by other opportunistic pathogens. IL-6 inhibitors should not be administered to patients when they have an active infection, including localized ones. Caution is needed when therapy is considered for patients with chronic or recurrent infections, those exposed to TB, patients with histories of serious or opportunistic infections, those who have resided in areas where TB is endemic, and patients with underlying conditions that predispose them to infections.
Gastrointestinal perforations have occurred in patients treated with IL-6 inhibitors. These most likely are complications of diverticulitis. Patients with RA have increased risk of diverticulitis. Most patients receiving IL-6 inhibitors who have developed perforations have also been receiving other agents that produce adverse gastrointestinal effects, including nonsteroidal anti-inflammatory drugs, corticosteroids, or methotrexate. Laboratory monitoring of IL-6 inhibitors should include complete blood counts, liver enzymes, and lipid profiles. In patients who develop neutropenia or thrombocytopenia, dose reduction or interruption may be needed. Dyslipidemias are managed with lipid-lowering therapies.
As with TNF-alpha inhibitors, live vaccines should not be used during IL-6 inhibitor therapy and for 3 months after discontinuation. Responses to inactivated vaccines may be blunted during IL-6 inhibitor therapy.8
Key Counseling Information
As noted in previous sections, several points can be emphasized during counseling of patients receiving IL-6 inhibitors:
- Advise patients to watch closely for signs or symptoms of infection. They should let the prescriber or pharmacist know if any of the following symptoms occur: fevers, chills, cough lasting more than 3 weeks, coughing up blood, unexplained weight loss, fatigue, or other indicators of infection as listed in the patient medication guide included with the product.
- Patients need regular blood tests to monitor for low neutrophil or platelet counts or increased liver function enzymes or cholesterol.
- Be sure patients know to report use of these agents to all health providers and that they know not to receive live vaccines.
- All products should be stored in the refrigerator and removed 30–60 minutes before use. Patients should allow the product to warm at room temperature and should not attempt to accelerate the warming process in any way, including by placing the product in warm water or microwaving.
RITUXIMAB
A chimeric monoclonal antibody, rituximab has a prolonged duration of activity on B cells that allows for dosing every 16–24 weeks in patients with RA. Rituximab treatment can lead to a rare but serious viral complication, progressive multifocal leukoencephalopathy (PML). Rituximab carries a black-box warning cautioning about PML and also serious, sometimes fatal infusion reactions, severe mucocutaneous reactions, and hepatitis B virus reactivation.8
Dosing and Administration
Rituximab acts by targeting the CD20 antigen of pre-B and mature B-lymphocytes. In patients with RA, this results in near depletion of peripheral B-lymphocytes within 2 weeks of the first dose. This effect persists for at least 6 months, allowing intermittent treatment based on disease symptoms.8
Rituximab is administered only as an intravenous infusion. Treatment begins with two 1000-mg infusions given 2 weeks apart. Patients should be pretreated with methylprednisolone and an antihistamine, as described below, before each infusion to reduce infusion reactions. This course is repeated 24 weeks later, or as soon as 16 weeks later based on clinical evaluation.8 In addition, rituximab is indicated only for use in combination with methotrexate.8
Adverse Reactions and Tolerability
The most common adverse reactions seen in patients with RA who are receiving rituximab (occurring in more than 10% of patients) are infusion-related reactions, upper respiratory infections, nasopharyngitis, urinary tract infection, and bronchitis. Infusion reactions occur in about one-third of patients and are characterized by fever, chills, rigors, pruritus, urticaria/rash, angioedema, sneezing, throat irritation, cough, and/or bronchospasm; hypertension or hypotension may accompany these symptoms.8 Finally, patients receiving rituximab should be premedicated 30 minutes before each dose with of intravenous methylprednisolone 100 mg and an oral antihistamine to avoid these reactions.24 For major infusion-related reactions, including anaphylaxis, drug discontinuation is essential.
The potentially fatal PML is a severe infection of the central nervous system caused by the John Cunningham virus (commonly referred to as JCV). Left untreated, this rapidly progressive disease can lead to severe disability or death.1 Of the biologics used to treat RA, PML has been associated only with rituximab therapy. Though it occurs infrequently and cases occur primarily in patients concomitantly using other immunosuppressive therapies, the manufacturer labeling for rituximab includes a black-box warning for PML.1 Monitor patients for symptoms of PML, including confusion, motor impairment, impaired coordination, and changes in speech, vision, or personality.16,24,35 The best outcomes with PML are found with early detection and prompt discontinuation of the offending agent.35
Warnings and Precautions
Other than PML and infusion reactions (described in the previous section), health professionals administering rituximab should be aware of several other warnings listed in product labeling8:
- Reactivation of HBV may occur in patients who are positive for core or surface antigens and in some patients whose infections appear to have resolved.
- Severe and sometimes fatal mucocutaneous reactions, including Steven-Johnsons syndrome and toxic epidermal necrolysis, have occurred in rituximab-treated patients. In patients with these reactions, rituximab should be discontinued. The safety of readministration in these patients has not been studied.
- As with other TNF and non-TNF biologics, serious infections can occur with rituximab therapy. These have included new or reactivated viral infections. Notably, there is no clear evidence of an association between the use of rituximab and an increased risk of tuberculosis.36
- Cardiovascular adverse events — including ventricular fibrillation, myocardial infarction, and cardiogenic shock — have occurred in patients treated with rituximab. The infusion should be discontinued in patients with life-threatening arrhythmias and monitored as needed in patients experiencing clinically significant arrhythmias.
- Administration of live vaccines in patients on rituximab has not been studied and is not recommended.
- Concomitant use of DMARDs other than methotrexate and use of other biologics with rituximab has not been well studied and should be attempted only with special attention on signs of infection.
- Rituximab should not be used in patients with RA unless they have not had adequate response to one or more TNF antagonists.
Key Counseling Information
A detailed patient medication guide is available for patients receiving rituximab therapy. Health professionals administering rituximab should review this document with patients, including its information about infusion reactions, severe skin and mouth reactions, viral infections, PML, and other possible adverse events.
PATIENT MONITORING
Efficacy
To ensure patients are benefiting from biologic therapy and avoid unnecessarily exposing patients to agents that can cause serious adverse reactions, disease activity must be monitored closely. Providers should follow-up with their anti-TNF-treated patients 3 months after initiating therapy and those taking non-TNF agents 6 months after the start of therapy, as these are the estimated times necessary to establish a response.9
Numerous disease activity measurement tools are available to providers. To narrow the field, the ACR released recommendations in which the group endorses the use of six disease activity scales (Table 5).37 These measurement tools are recommended based on their reliability, validity, and responsiveness to change in disease activity. For all of these measures, providers can compare patient values with predefined scales to determine disease severity, including whether the patient is in clinical remission.37
| Table 5. Tools Recommended by the American College of Rheumatology for Measuring Disease Activity in Patients With Rheumatoid Arthritis |
| Tools |
Methods of Administration |
Required Components |
Scoring Outputs |
| Physician Joint Count |
Patient Global VAS |
Provider Global VAS |
Pain |
| PAS |
Patient questionnaire |
|
✓ |
|
✓ |
0–10 |
| PAS-II |
Patient questionnaire |
|
✓ |
|
✓ |
0–10 |
| RAPID-3 |
Patient questionnaire |
|
✓ |
|
✓ |
0–10 |
| CDAI |
Provider item; patient item |
✓ |
✓ |
✓ |
|
0–76 |
| DAS28(ESR or CRP) |
Provider assessment; patient item; lab |
✓ |
✓ |
|
|
0–9.4 |
| SDAI |
Provider assessment; patient item; lab |
✓ |
✓ |
✓ |
|
0–86 |
Source: Adapted from Reference 37.
Abbreviations used: CDAI – Clinical Disease Activity Index; DAS28(ESR or CRP) – Disease Activity Score with 28-joint counts with erythrocyte sedimentation rate or C-reactive protein; PAS – Patient Activity Scale; RAPID-3 – Routine Assessment of Patient Index Data with three measures; SDAI – Simplified Disease Activity Index; VAS – Visual Analog Scale. |
Safety
While biologic DMARDs have been shown to be effective for the treatment of RA, concerns about their safety may lead to hesitation on the part of the provider and patient. However, with proper monitoring and management of adverse events, the risks can be mitigated. Monitoring for safety is especially important when starting or resuming biologic therapy, when increasing the dose significantly, and in patients receiving multiple immunosuppressants.21
DRUG COST
All of the biologic DMARDs are priced similarly, costing thousands of dollars each month. For this reason, health insurance plans will typically "prefer" a limited number of these agents to secure more favorable pricing from manufacturers. The decision to prefer certain agents is based on safety and cost-effectiveness data. Given the number of relatively therapeutically equivalent options, the preferred biologics will likely vary between payers based on manufacturer rebates. By selecting the most clinically appropriate agent of those covered by patients' insurance plans, providers can help ensure patients stay adherent to therapy and do not experience financial hardship due to the monumental cost of these medications. Further, providers can work with third-party payers to get coverage for the most appropriate medications if the payers' preferred alternatives are unsuitable for certain patients.
Comparative Efficacy Evidence
While biologic DMARDs have been established as a sound therapeutic option for patients with moderately to severely active RA, few data exist that adequately compare the effectiveness of these agents. Much of the evidence is indirect, comparing the findings of numerous trials to one another. Results from indirect comparisons must be interpreted with caution given the innate differences in populations and methodology between trials.
From the significant amount of indirect comparative evidence available, including numerous meta-analyses and systematic reviews, several assumptions can be made. Though the biologics have persistently been recognized as more effective therapeutic options than placebo,14,38 there is still limited head-to-head data comparing biologic treatment for RA. In a meta-analysis published in 2016, all biologic treatments for RA, except for etanercept, had a higher response rate when taken with a traditional DMARD. Higher disease response rates were seen with higher doses of biologic DMARDs, suggesting a dose-dependent relationship. When given as monotherapy, biologic agents were ranked from the most to the least effective as follows: certolizumab, etanercept, tocilizumab/abatacept (one is not more effective than the other), adalimumab. The ranking of biologic treatment differs slightly when the biologic agent is given in combination with another drug. When given as an add-on to existing therapy, the most effective to the least effective biologics were as follows: certolizumab, tocilizumab, anakinra, rituximab, golimumab/infliximab/abatacept, and adalimumab/etanercept.10
The primary takeaway point is that there is a very limited body of head-to-head research with which to work and the conclusions drawn above are not absolute. Indirect data may be useful to highlight areas requiring more research, but until direct comparison data become available, the results of the previously mentioned systematic reviews and meta-analyses should be interpreted with caution when implementing findings in clinical practice.
Biosimilars
Biosimilars are now reaching the U.S. market. A biosimilar is a highly similar drug product to an innovator biologic agent, also known as a reference product, but is not identical to that agent.41,42 Biosimilars have essentially the same active ingredient and mechanism of action and are intended to treat the same disease states as their reference products.41,43 However, they have minor differences secondary to their intricate chemical structures and manufacturing processes that may cause the biosimilar to have a different safety and/or efficacy profile than the originator product.43
Biosimilars have been on the market in Europe since 2006. In the U.S., the Biologics Price Competition and Innovation Act (BPCI) of 2009, passed as part of the Patient Protection and Affordable Care Act, streamlined the process for biosimilar entry to the market. Biosimilars differ from traditional drugs in a number of ways. First, biologic drugs are significantly larger in size and have a more complex structure as opposed to traditional small drug compounds.41,42 Further, due to the unstable nature of these compounds, they typically require special storage and handling and are more likely to be immunogenic than small drug compounds.42 Biologics are made through a more complex manufacturing process that is less predictable than that of traditional agents.41 Thus, manufacturers of generic small drug compounds can more easily replicate the predictable chemical reactions required to create an identical product.42
While the BPCI created an abbreviated approval process for biosimilars, the path to approval still requires rigorous testing. Unlike traditional generics that simply have to show bioequivalence for approval, biosimilar manufacturers must show the product is "highly similar to the original innovator product, notwithstanding minor differences in clinically inactive components," and must provide clinical research to demonstrate no substantial differences in safety, purity, and potency compared with the reference product.44
By introducing competition to the market, biosimilars are expected to reduce biologic prices by approximately 35% — reducing costs by an estimated $44.2 billion over the next 10 years.45 The Congressional Budget Office predicts a more considerable average price reduction of about 40%. Based on experience from biosimilars in Europe, the estimated savings upon biosimilar entry to the market is often 25% or more. Though this would substantially reduce the costs of biologics, this is significantly lower than the average 50% to 80% cost reduction of small-molecule generics on the U.S. market.
The anti-TNF class has the largest financial opportunity of all biologics, accounting for more than 20% of the savings projected above.45 The first biosimilar indicated for the treatment of RA approved by FDA was Inflectra (infliximab-dyyb) in April 2016.46 Since then, seven additional biosimilars for this condition have been approved (Table 6). In general, biosimilars have been shown to be as safe and effective in clinical studies leading to approval.47-53 However, real world experience is lacking and further data are needed to confirm these findings.50-52
| Table 6. List of FDA-Approved Biosimilars Indicated for Rheumatoid Arthritis |
| Brand Name |
Generic Name |
Originator Product |
Date of FDA Approval |
| Inflectra |
infliximab-dyyb |
Remicade (infliximab) |
April 2016 |
| Erelzi |
etanercept-szzs |
Enbrel (etanercept) |
August 2016 |
| Amjevita |
adalimumab-atto |
Humira (adalimumab) |
September 2016 |
| Renflexis |
infliximab-adba |
Remicade (infliximab) |
May 2017 |
| Cyltezo |
adalimumab-adbm |
Humira (adalimumab) |
August 2017 |
| Ixifi |
infliximab-qbtx |
Remicade (infliximab) |
December 2017 |
| Hyrimoz |
adalimumab-adaz |
Humira (adalimumab) |
October 2018 |
| Truxima |
rituximab-abbs |
Rituxan (rituximab) |
November 2018 |
Source: Adapted from Reference 46.
Abbreviation used: FDA, Food and Drug Administration |
While these products are approved for use in the United States, several barriers to uptake still remain. First, not all of these products are currently commercially available. For example, Amgen has no intention to launch Amjevita (adalimumab-atto) before 2023, while Pfizer has announced Ixifi (infliximab-qbtx) will not be marketed in this country.54 Whatever the reasons for these delayed launches, health care professionals should expect delays in the anticipated cost-saving potential of biosimilars. Additionally, all biosimilars in the U.S. currently lack an interchangeability designation.55 This limits the pharmacist’s ability to swap an originator product for a biosimilar at the point of sale without the intervention of the prescriber. As more biosimilars become commercially available and gain the interchangeability designation, the cost savings of biosimilars will become more apparent.
CONCLUSION
Biologic agents have strong evidence to support their use with tolerable safety profiles in patients with moderate-to-severe RA. While more data are emerging, no clear evidence currently suggests that one biologic agent is superior to the alternatives in terms of efficacy or safety. Much of the efficacy and safety data involve indirect comparisons, which must be interpreted with caution due to the inevitable variation in study designs and populations.
The primary recommendation of the ACR RA guideline is to choose an agent based on its mechanism of action. By simply categorizing the biologics into three groups (anti-TNF biologics, non-TNF biologics, and tofacitinib), the guideline subtly highlights the lack of direct comparative efficacy and safety data. Patient-specific factors — such as dosing, other underlying diseases, and patient preference — can then be used to choose the most appropriate agent for each individual.
However, the provider's role does not end with the selection of a biologic agent. RA is associated with a decreased quality of life and providers, including pharmacists, can help patients control this complex condition in a variety of ways after initiating biologic therapy. Through proper follow-up and management of adverse reactions, patients can experience prolonged benefit with therapy and subsequently all the advantages associated with decreased disease activity.
REFERENCES
- Thaler KJ, Gartlehner G, Kien C, et al. Drug class review: targeted immune modulators: final update 3 report. Portland, OR: Oregon Health & Science University; 2012. http://www.ncbi.nlm.nih.gov/books/NBK110098/
- Klarenbeek NB, Kerstens PJ, Huizinga TW, Dijkmans BA, Allaart CF. Recent advances in the management of rheumatoid arthritis. BMJ. 2010;341:c6942.
- Wasserman AM. Diagnosis and management of rheumatoid arthritis. Am Fam Physician. 2011;84(11):1245-1252.
- Rheumatoid arthritis in adults: management. National Institute for Health and Care Excellence (NICE). 2018. Accessed at https://www.nice.org.uk/guidance/ng100/resources/rheumatoid-arthritis-in-adults-management-pdf-66141531233989, April 23, 2019.
- Rindfleisch JA, Muller D. Diagnosis and management of rheumatoid arthritis. Am Fam Physician. 2005 Sep 15;72(6):1037-1047.
- Boot CRL, de Wind A, van Vilsteren M, et al. One-year predictors of presenteeism in workers with rheumatoid arthritis: disease-related factors and characteristics of general health and work. J Rheumatol. 2018;45(6):766-770.
- Williams RO. What have we learned about the pathogenesis of rheumatoid arthritis from TNF-targeted therapy? ISRN Immunology. 2012.
- S. Department of Health & Human Services. Drugs@FDA: FDA approved drug products. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/
- Singh JA, Saag KG, Bridges SL Jr., et al. 2015 American College of Rheumatology guideline for the treatment of rheumatoid arthritis. Arthritis Rheum. 2016;68(1):1–26.
- Tvete IF, Natvig B, Gåsemyr J, et al. Comparing effects of biologic agents in treating patients with rheumatoid arthritis: a multiple treatment comparison regression analysis. PLoS One. 2015 Sep 10;10(9):e0137258.
- Aaltonen KJ, Virkki LM, Malmivaara A, et al. Systematic review and meta-analysis of the efficacy and safety of existing TNF blocking agents in treatment of rheumatoid arthritis. PLoS One. 2012;7(1):e30275.
- Emery P, Fleischmann RM, Moreland LW, et al. Golimumab, a human anti-tumor necrosis factor alpha monoclonal antibody, injected subcutaneously every four weeks in methotrexate-naive patients with active rheumatoid arthritis: twenty-four-week results of a phase III, multicenter, randomized, double-blind, placebo-controlled study of golimumab before methotrexate as first-line therapy for early-onset rheumatoid arthritis. Arthritis Rheum. 2009;60(8):2272-2283.
- Edwards JC, Szczepanski L, Szechinski J, et al. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004;350(25):2572-2581.
- Buckley F, Finckh A, Huizinga TW, et al. Comparative efficacy of novel DMARDs as monotherapy and in combination with methotrexate in rheumatoid arthritis patients with inadequate response to conventional DMARDs: a network meta-analysis. J Manag Care Spec Pharm. 2015;21(5):409-423.
- Aaltonen KJ, Joensuu JT, Virkki L, et al. Rates of serious infections and malignancies among patients with rheumatoid arthritis receiving either tumor necrosis factor inhibitor or rituximab therapy. J Rheumatol. 2015;42(3):372-378.
- Selmi C, Ceribelli A, Naguwa SM, et al. Safety issues and concerns of new immunomodulators in rheumatology. Expert Opin Drug Saf. 2015;14(3):389-399.
- Singh JA, Cameron C, Noorbaloochi S, et al. Risk of serious infection in biological treatment of patients with rheumatoid arthritis: a systematic review and meta-analysis. Lancet. 2015;386(9990):258-265.
- Singh JA, Wells GA, Christensen R, et al. Adverse effects of biologics: a network meta-analysis and Cochrane overview. Cochrane Database Syst Rev. 2011; (2):CD008794.
- Michaud TL, Rho YH, Shamliyan T, et al. The comparative safety of tumor necrosis factor inhibitors in rheumatoid arthritis: a meta-analysis update of 44 trials. Am J Med. 2014;127(12):1208-1232.
- Knittle K, Maes S, de Gucht V. Psychological interventions for rheumatoid arthritis: examining the role of self-regulation with a systematic review and meta-analysis of randomized controlled trials. Arthritis Care Res (Hoboken). 2010;62(10):1460-1472.
- Rubbert-Roth A. Assessing the safety of biologic agents in patients with rheumatoid arthritis. Rheumatology (Oxford). 2012;51 Suppl 5:v38-47.
- Kroesen S, Widmer AF, Tyndall A, Hasler P. Serious bacterial infections in patients with rheumatoid arthritis under anti-TNF-a therapy. Rheumatology (Oxford). 2003;42:617-621.
- Kumar P, Banik S. Pharmacotherapy options in rheumatoid arthritis. Clin Med Insights Arthritis Musculoskelet Disord. 2013;6:35-43.
- Nanau RM, Neuman MG. Safety of anti-tumor necrosis factor therapies in arthritis patients. J Pharm Pharm Sci. 2014;17(3):324-361.
- Parekh MJ, Schluger NW. Treatment of latent tuberculosis infection. Ther Adv Respir Dis. 2013;7(6):351-356.
- Sterling TR, Villarino ME, Borisov AS, et al. Three months of rifapentine and isoniazid for latent tuberculosis infection. N Engl J Med. 2011;365(23):2155-2166.
- Lebrec H, Ponce R, Preston BD, et al. Tumor necrosis factor, tumor necrosis factor inhibition, and cancer risk. Curr Med Res Opin. 2015;31(3):557-574.
- Park HJ, Ranganathan P. TNF-alpha antagonism and cancer risk in rheumatoid arthritis: is continued vigilance warranted? Discov Med. 2012;13(70):229-234.
- Solomon DH, Reed GW, Kremer JM, et al. Disease activity in rheumatoid arthritis and the risk of cardiovascular events. Arthritis Rheumatol. 2015;67(6):1449-1455.
- Greenberg JD, Kremer JM, Curtis JR, et al. Tumour necrosis factor antagonist use and associated risk reduction of cardiovascular events among patients with rheumatoid arthritis. Ann Rheum Dis. 2011;70(4):576-582.
- Zhao Q, Hong D, Zhang Y, et al. Association between anti-TNF therapy for rheumatoid arthritis and hypertension: a meta-analysis of randomized controlled trials. Medicine (Baltimore). 2015;94(14):e731.
- Lequerré T, Vittecoq O, Klemmer N, et al. Management of infusion reactions to infliximab in patients with rheumatoid arthritis or spondyloarthritis: experience from an immunotherapy unit of rheumatology. J Rheumatol. 2006;33(7):1307-1314.
- Rajakulendran S, Deighton C. Adverse dermatological reactions in rheumatoid arthritis patients treated with etanercept, an anti-TNF-alpha drug. Curr Drug Saf. 2006;1(3):259-264.
- Hua C, Barnetche T, Combe B, Morel J. Effect of methotrexate, anti-tumor necrosis factor α, and rituximab on the immune response to influenza and pneumococcal vaccines in patients with rheumatoid arthritis: a systematic review and meta-analysis. Arthritis Care Res (Hoboken). 2014 Jul;66(7):1016-1026.
- Borie D, Kremer JM. Considerations on the appropriateness of the John Cunningham virus antibody assay use in patients with rheumatoid arthritis. Semin Arthritis Rheum. 2015.
- Chen YM, Chen HH, Lai KL, et al. The effects of rituximab therapy on released interferon-γ levels in the QuantiFERON assay among RA patients with different status of Mycobacterium tuberculosis infection. Rheumatology (Oxford). 2013;52(4):697-704.
- Anderson J, Caplan L, Yazdany J, et al. Rheumatoid arthritis disease activity measures: American College of Rheumatology recommendations for use in clinical practice. Arthritis Care Res. 2012;64(5):640-647.
- Orme ME, MacGilchrist KS, Mitchell S, et al. Systematic review and network meta-analysis of combination and monotherapy treatments in disease-modifying antirheumatic drug-experienced patients with rheumatoid arthritis: analysis of American College of Rheumatology criteria scores 20, 50, and 70. Biologics. 2012;6:429-464.
- Jansen JP, Buckley F, Dejonckheere F, Ogale S. Comparative efficacy of biologics as monotherapy and in combination with methotrexate on patient reported outcomes (PROs) in rheumatoid arthritis patients with an inadequate response to conventional DMARDs – a systematic review and network meta-analysis. Health Qual Life Outcomes. 2014;12:102.
- Schiff M, Keiserman M, Codding C, et al. Efficacy and safety of abatacept or infliximab vs placebo in ATTEST: a phase III, multi-centre, randomised, double-blind, placebo controlled study in patients with rheumatoid arthritis and an inadequate response to methotrexate. Ann Rheum Dis. 2008;67(8):1096-1103.
- Li E, Ramanan S, Green L. Pharmacist substitution of biological products: issues and considerations. J Manag Care Spec Pharm. 2015;21(7):532-539.
- Camacho LH, Frost CP, Abella E, et al. Biosimilars 101: considerations for U.S. oncologists in clinical practice. Cancer Med. 2014;3(4):889-899.
- van de Vooren K, Curto A, Garattini L. Biosimilar versus generic drugs: same but different? Appl Health Econ Health Policy. 2015;13(2):125-127.
- Biologics Price Competition and Innovation Act of 2009. Accessed at https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/ucm216146, April 23, 2019.
- Mulcahy AW, Predmore Z, Mattke S. The cost savings potential of biosimilar drugs in the United States. RAND Corporation. Accessed at http://www.rand.org/pubs/perspectives/PE127.html Published 2014, April 23, 2019.
- S. Food and Drug Administration. Biosimilar product Information. Updated 2018 Nov 28. Accessed at https://www.fda.gov/drugs/developmentapprovalprocess/howdrugsaredevelopedandapproved/approvalapplications/therapeuticbiologicapplications/biosimilars/ucm580432.htm, April 23, 2019.
- Baji P, Péntek M, Czirják L, et al. Efficacy and safety of infliximab-biosimilar compared to other biological drugs in rheumatoid arthritis: a mixed treatment comparison. Eur J Health Econ. 2014;15(Suppl 1):53-64.
- McKeage K. A review of CT-P13: an infliximab biosimilar. BioDrugs. 2014;28(3):313-321.
- Bae SC, Lee YH. Comparative efficacy and safety of biosimilar-infliximab and originator-infliximab in combination with methotrexate in patients with active rheumatoid arthritis: a meta-analysis of randomized controlled trials. Int J Rheum Dis. 2018;21(5):922-929.
- Chadwick L, Zhao S, Mysler E, Moots RJ. Review of biosimilar trials and data on etanercept in rheumatoid arthritis. Curr Rheumatol Rep. 2018;20(12):84.
- Zhao S, Chadwick L, Mysler E, Moots RJ. Review of biosimilar trials and data on adalimumab in rheumatoid arthritis. Curr Rheumatol Rep. 2018;20(10):57.
- McKinnon RA, Cook M, Liauw W, et al. Biosimilarity and interchangeability: principles and evidence: a systematic review. BioDrugs. 2018;32(1):27-52.
- Inotai A, Prins CPJ, Csanádi M, et al. Is there a reason for concern or is it just hype? – A systematic literature review of the clinical consequences of switching from originator biologics to biosimilars. Expert Opin Biol Ther. 2017;17(8):915-926.
- D’Arrigo T. Delayed launches and patent litigation limit biosimilar availability. Pharmacy Today. 2018;24(10):6.
- Rychlick N. Biosimilars for rheumatoid arthritis: don't count them out quite yet. Manag Care. 2018;27(1):29.
Back to Top