
Expired activity
Please go to the PowerPak
homepage and select a course.
Role of Specialty Pharmacists in Optimizing Management of Patients with PAH
INTRODUCTION
Pulmonary arterial hypertension (PAH) is a progressive disease characterized by increases in pulmonary vascular resistance which ultimately lead to right ventricular (RV) failure and death.1 However, significant improvements in the clinical outcomes of PAH have been realized by recent advances in pharmacotherapy. Consequently, specialty pharmacists can play an integral role in the delivery of optimal care for patients with PAH.
Epidemiology, pathogenesis and patient registries
PAH is an orphan disease state with an overall incidence of 15 per million people.2 It is also a disease that predominantly affects younger female patients with recent U.S. registry data revealing a mean age of 50 ± 14 years among a population that is 80% female.3 Median survival was found to be 7 years for patients enrolled between 2006 and 2007 into the U.S. Registry to Evaluate Early and Long-Term PAH Disease Management (REVEAL).4 This represents a marked improvement compared to a median survival of only 2.8 years per registry data from the U.S. National Institute of Health Registry collected in the 1980's.5 This change parallels the increased availability of effective treatment options over the past two decades.
The pathogenesis of PAH is complex and multifactorial. However, the central features of the disease pathophysiology include vasoconstriction, vascular wall remodeling, and in situ thrombosis.1 The culmination of each of these leads to an increase in vascular resistance. PAH is characterized by vasculopathy which mainly affects small pulmonary arteries through intimal hyperplasia, medial hypertrophy, adventitial proliferation, inflammation, thrombosis, and formation of plexiform lesions.6 Excessive vasoconstriction also plays a role in the pathogenesis. A dysregulation and imbalance of vasodilators (i.e., nitric oxide, prostacyclins) and vasoconstrictors (i.e., thromboxane A2, endothelin-1) within the vascular endothelium and smooth muscle provide a construct for the major pathways and therapeutic targets of PAH.1
Nitric oxide (NO) is a potent vasodilator and also inhibits platelet activation and vascular smooth muscle cell proliferation.6 A deficiency of nitric oxide (NO) is a result of impaired synthesis and signaling via the NO-soluble guanylate cyclase (sGC)-cyclic guanosine monophosphate (cGMP) pathway.7 Nitric oxide activates its molecular target, sGC, which in turn leads to increases in cGMP production. The degradation of cGMP is regulated by the enzyme, phosphodiesterase-type 5 (PDE-5).6 Prostacyclin (prostaglandin I2 or PGI2) and thromboxane A2 are major metabolites of arachidonic acid metabolism, which have significant effects on pulmonary arterial vascular smooth muscle. Whereas prostacyclin is a potent vasodilator, an inhibitor of platelet activation, and has antiproliferative properties, thromboxane A2 exhibits the opposite effects (i.e., vasoconstriction, stimulation of platelet activation, and promotion of cell proliferation).6 In PAH, there is an excess of thromboxane A2 and a deficiency of prostacyclin. Endothelin-1 (ET-1) is a potent vasoconstrictor and mitogen that exerts its effects in the pulmonary vascular smooth muscle via endothelin type A (ETA) and endothelin type B (ETB) receptors.7 In PAH, ET-1 concentrations are elevated and its clearance is impaired.6
Risk factors, presentation and diagnosis (Evaluating severity and determinants of risk)
The onset of PAH may be insidious and the presentation can be very nonspecific. Symptoms may include dyspnea, fatigue, exercise intolerance, chest pain, fluid retention, and less commonly syncope.6 Consequently, careful patient assessment is warranted and should include comprehensive testing using echocardiography, pulmonary function tests, sleep studies, serologies, ventilation-perfusion scanning, and computed tomography scans of the chest to evaluate for possible underlying causes. When pulmonary hypertension (PH) is suspected, a right heart catheterization (RHC) is required to confirm the diagnosis. Specific hemodynamic criteria must also be met for diagnosis and includes a mean pulmonary arterial pressure of ≥ 25 mm Hg accompanied by a pulmonary capillary wedge pressure (PCWP) of ≤ 15 mm Hg.6 The latter is necessary to exclude the presence of passive increases in pulmonary pressures on the basis of increased preload due to left-sided heart disease.
The severity of patient symptoms is determined by the World Health Organization (WHO) functional classification: class I (symptoms elicited at levels of exertion that would limit normal Individuals), class II (symptoms on ordinary exertion), class III (symptoms on less-than-ordinary exertion), and class IV (symptoms at rest). It is noteworthy that 56% of the PAH population in the U.S. REVEAL registry had WHO functional class (FC) III or IV symptoms.4
Overall risk assessment for determining prognosis and treatment approach in PAH is based on a constellation of clinical variables (Table 1), with RV function being a major predictor of functional capacity and outcome.6 From the contemporary REVEAL registry data, several patient-specific factors were identified to either increase or decrease the risk of death among patients with PAH. Factors associated with increased mortality include portal hypertension and connective tissue disease associated with PAH, family history of PAH, renal insufficiency, males > 60 years of age, WHO FC III-IV symptoms, resting systolic blood pressure < 110 mm Hg, heart rate > 92 beats per minute at rest, 6-minute walk distance (6MWD) < 165 meters, brain-type natriuretic peptide (BNP) > 180 pg/mL, pulmonary vascular resistance > 32 Wood units, mean right atrial pressure (RAP) > 20 mm Hg, presence of pericardial effusion, and percent predicted diffusion capacity of carbon dioxide (DLCO) ≤ 32%. Conversely, factors associated with improved survival include WHO FC I, 6MWD ≥ 440 meters, BNP < 50 pg/mL, and DLCO ≥ 80%.8
| Table 1. Risk Assessment in Patients with Pulmonary Arterial Hypertension16,17 |
| Risk Factor |
Lower Risk |
Higher Risk |
| |
|
|
| Evidence of RV failure |
No |
Yes |
| Symptom progression |
Gradual |
Rapid |
| Functional class |
II, III |
IV |
| 6-minute walk distance |
> 400 meters |
< 300 meters |
| Exercise testing |
Peak VO2 > 10.4 mL/kg/min |
Peak VO2 < 10.4 mL/kg/min |
| Echocardiogram |
Minimal RV dysfunction |
Pericardial effusion; RV enlargement/dysfunction; right atrial enlargement |
| Hemodynamics |
RAP < 10 mm Hg; CI > 2.5 L/min/m2 |
RAP > 20 mm Hg; CI < 2 L/min/m2 |
| BNP |
Minimal elevation |
Significant elevation |
| |
| BNP = B-type natriuretic peptide; CI = cardiac index; RAP = right atrial pressure; RV = right ventricular; VO2 = peak oxygen consumption |
PH Classification and Implications for Targeted Treatments
The clinical classification of PH (Table 2) was most recently updated during the Fifth World Symposium on Pulmonary Hypertension (WSPH) in 2013. The WHO classification places PH patients into four distinct groups based on shared pathologic features, similarities in hemodynamics, and management strategies.9 The majority of pharmacotherapeutic treatment options are approved for patients with Group 1 disease. In the U.S. REVEAL registry, patients with Group 1 disease were further classified into three major subtypes: associated PAH (50.7%), idiopathic PAH (46.2%), and familial PAH (2.7%).3 Patients with the associated pulmonary arterial hypertension (APAH) subtype were yet further divided into subgroups based on cause, which included connective tissue disease (49.9%), congenital heart disease (19.5%), portal hypertension (10.6%), drug/toxin-induced (10.5%), HIV associated (4%), and other (5.5%).4
| Table 2. Clinical Classification of Pulmonary Arterial Hypertension11 |
World Health
Organization Group |
Description |
Subcategories |
|
1
|
Pulmonary arterial hypertension
|
Idiopathic
Heritable (BMPR2, ALK-1, ENG, SMAD9, CAV1, KCNK3)
Drug and toxin-induced (see Table 2)
Associated with connective tissue disorders
Associated with HIV infection
Associated with portal hypertension
Associated with congenital heart diseases
Associated with schistosomiasis
Pulmonary veno-occlusive disease and/or pulmonary capillary hemangiomatosis
Persistent pulmonary hypertension of the newborn
|
|
2
|
Pulmonary venous hypertension due to left heart disease
|
Left ventricular systolic dysfunction (heart failure with reduced ejection fraction)
Left ventricular diastolic dysfunction (heart failure with preserved ejection fraction)
Valvular disease
Congenital/acquired inflow/outflow tract obstruction and congential cardiomyopathies
|
|
3
|
Pulmonary hypertension due to lung diseases and/or hypoxia
|
Chronic obstructive pulmonary disease
Interstitial lung disease
Other pulmonary diseases with mixed restrictive and obstructive pattern
Sleep-disordered breathing (i.e., obstructive sleep apnea)
Alveolar hypoventilation disorders
Chronic exposure to high altitude
Developmental lung diseases
|
|
4
|
Chronic thromboembolic pulmonary hypertension (CTEPH)
|
|
|
5
|
Pulmonary hypertension with unclear multifactorial mechanisms
|
Hematologic disorders (chronic hemolytic anemia, myeloproliferative disorders, splenectomy)
Systemic disorders (sarcoidosis, pulmonary histiocytosis, lymphangioleiomyomatosis)
Metabolic disorders (glycogen storage disease, Gaucher disease, thyroid disorders)
Others (tumoral obstruction, fibrosing mediastinitis, chronic renal failure, segmental pulmonary hypertension)
|
| |
| BMPR2 = bone morphogenic protein receptor type II; CAV1 = caveolin-1; ENG = endoglin; HIV = human immunodeficiency virus; KCNK3 = Potassium channel subfamily K member 3; SMAD9 = Mothers against decapentaplegic homolog 9; ALK-1 = activin receptor-like kinase-1 |
Connective tissue diseases represent the most common cause of associated PAH. Half of these disorders are attributed to scleroderma, also referred to as systemic sclerosis and formerly known as calcinosis, Raynaud's phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasias (CREST) syndrome.10 The prevalence of PAH among people with scleroderma is between 7 and 12% and is a major cause of mortality.11,12 Other types of connective tissue disorders associated with PAH include systemic lupus erythematosus, Sjogren's syndrome, rheumatoid arthritis, and mixed connective tissue disease.9,12
Idiopathic PAH, formerly known as primary PH, is the second most common subset of patients with Group 1 disease. In familial PAH, genetic mutations can be detected in 80% of families with multiple cases of PAH.3,11 The most common mutation is in the bone morphogenetic protein receptor type 2 (BMPR2), a member of the tumor growth factor-beta superfamily.11 Other less commonly seen genetic mutations have been described. However, no detectable changes in disease-associated genes can be identified in 20% of patients.11 The pathology of idiopathic disease and PAH associated with connective tissue disorders is similar. Therefore, the treatments are generally similar but the relative benefits are less pronounced in patients with connective tissue disease.11,12
A summary of drugs and toxins that can induce PAH and the likelihood of association are outlined in Table 3. Historically, anorexigens such as fenfluramine and dexfenfluramine were found to be associated with an increased risk for developing PAH based on their ability to increase serotonin release and block serotonin reuptake.6 Selective serotonin reuptake inhibitors (SSRIs) have similarly been implicated in the development of persistent PH in newborn infants whose mothers were exposed to these medications in the later stages of pregnancy.11,13 An association between SSRI exposure and PH in adults remains unclear. Amphetamines have also been shown to be associated with PAH, and caution is therefore warranted with the use of related derivatives, such as phentermine, topiramate, methylphenidate, and ropinirole.11 Another novel class of medications known as the tyrosine kinase inhibitors have recently been investigated for their potential association with PAH.9 At this time, dasatinib appears to have the highest association with the development of PAH compared to other TKIs.9
| Table 3. Drug and Toxin-Induced Pulmonary Arterial Hypertension11 |
| Definite |
Likely |
Possible |
Unlikely |
|
Aminorex
Fenfluramine
Dexfenfluramine
Toxic rapeseed oil
Benfluorex
SSRIs†
|
Amphetamines
L-tryptophan
Methamphetamines
Dasatinib
|
Cocaine
Phenylpropanolamine St. John's wort
Chemotherapeutic agents
Interferon α and β
Amphetamine-like drugs
|
Oral contraceptives
Estrogen
Cigarette smoking
|
| |
SSRIs = selective serotonin reuptake inhibitors
†SSRIs have been determined to be a risk factor for the development of persistent pulmonary hypertension of the newborn in pregnant women exposed to SSRIs after 20 weeks of gestation. |
Treatment goals and patient monitoring
Several clinical variables are beneficial in establishing baseline risk and can be useful in helping to guide selection of initial drug therapy. Reassessment of these parameters can also help to determine ongoing response to therapy. These variables are most useful when combined to form composite goals. Examples of risk prediction variables in PAH are summarized below along with specific goals for each as part of a goal-oriented approach to therapy. This treat-to-target strategy utilizes predetermined treatment goals and refinement of treatment strategy if those goals are not achieved.14
The 6MWD is a non-invasive test that is simple, reproducible, and correlates with hemodynamics and survival.7 Consequently, this test has been used as the primary endpoint for a majority of key clinical trials in PAH.15 Assessment of hemodynamics is also critical for monitoring response to therapy, with right atrial pressure (RAP) and cardiac index (CI) measurements being better prognostic variables and pulmonary artery pressure (PAP) being less reliable due to its declination in the context of RV failure.15 Other prognostic variables of importance include RV function (as assessed by echocardiography and/or cardiac magnetic resonance imaging [cMRI]), FC, cardiopulmonary exercise testing, and biomarkers (i.e., BNP).15
Currently recommended treatment goals include attainment of WHO FC I or II, 6MWD > 380 to 440 meters, normalization of RV function (RAP < 8 mm Hg and CI > 2.5 to 3.0 L/min/m2), normal or near-normal RV size and function by echocardiography or cMRI, normalization of biomarkers (BNP), and peak oxygen consumption (VO2) > 15 mL/min/kg.15
Pharmacotherapeutic agents in PAH
Few orphan diseases have seen such rapid evolution in available therapy. Over the last 2 decades, approximately 14 treatment options for PAH came to market whereas options were previously limited to supportive care or calcium channel blockers in a small number of patients. New research continues in the areas of combination therapies and innovative pathways in the hopes of further impacting the underlying disease and slowing or preventing deterioration. The therapeutic approach has evolved as well with updates in guidelines that place more emphasis on specific risk factors and disease severity to help guide initial and ongoing treatment.
Table 4 summarizes the current treatment options for PAH. These therapies are only approved for use in patients diagnosed with Group 1 PAH with the exception of riociguat, which also possesses an indication for chronic thromboembolic pulmonary hypertension (CTEPH). In fact, evidence for use in other types of PH is marginal at best and potentially harmful in some circumstances; therefore, it is essential for patients to be referred to expert centers prior to starting drug therapy. Treatments for PAH target the underlying pathobiology of the disease along one of three known pathways, including endothelin, nitric oxide, and prostacyclin.18,19
| Table 4: PAH Targeted Therapies20-32 |
| How Supplied |
Administration |
FC |
Dose |
Properties |
CI/P |
Misc. |
|
Epoprostenol
(generic, Flolan®, Veletri®*
0.5 mg, 1.5 mg lyophilized powder for reconstitution)
|
Continuous intravenous infusion via
infusion pump
Requires tunneled CVC
Flolan requires use of ice packs during infusion
|
III, IV
|
Initiated at 2 ng/kg/min and titrated based on response
Ongoing: 1-2 ng/kg/min every 1 to 2 weeks
|
T ½ < 6 min
Temp and light sensitive
Reconstituted stability dependent on conc. And formulation
Rapidly hydrolyzed in the blood
|
CHF due to severe LVD
Avoid abrupt withdrawals or interruption in infusion: may result in rebound PH or death.
|
Initiated in controlled setting such as a hospital
Monitor for signs of BSI
Only available through LDD
|
|
Treprostinil
(Remodulin® 1 mg/ml, 2 mg/ml, 5 mg/ml, 10 mg/ml in 20 ml vials)
(Tyvaso® for inhalation 0.6 mg/ml in 2.9 ml ampules)
(Orenitram® 0.125 mg, 0.25 mg, 1 mg and 2.5 mg ER tablets)
|
Continuous IV or SubQ infusion via infusion pump
IV requires tunneled CVC
Intermittent inhalation via dedicated inhalation device
Oral extended release osmotic tablets
|
Infused: II-IV
Inhaled: primarily III
Oral: II, III
|
Initiated at 1.25 ng/kg/min and titrated based on response
Ongoing: 1.25 ng/kg/min every week or as tolerated
Inhaled: start at 3 breaths QID, titrated to goal 9 breaths QID. May use doses up to 15 breaths QID
Oral: Initial: 0.25 mg BID or 0.125 mg TID, titrate every 3 to 4 days
|
T ½ ~ 4 hours. Metabolized by CYP 2C8.
Diluted: 48-hour infusion duration
Undiluted: 72 hour infusion duration
Inhaled: protect ampules from light during storage. Once opened: discard remaining solution after 24 hours
Oral: Food increases bioavailability
|
CHF due to severe LVD
Avoid abrupt withdrawals or interruption in infusion: may result in rebound PH or death.
Oral: Diverticulitis
Severe hepatic impairment
Avoid alcohol
|
Infused: Initiated in controlled setting such as a hospital
Monitor for signs of BSI
Inhaled: One inhaled ampule provides multiple doses/day
Only available through LDD
|
|
Iloprost
(Ventavis® 10 mcg/ml and 20 mcg/ml unit dose ampules)
|
Intermittent inhalation via dedicated inhalation device
|
III, IV
|
Initial: 2.5 mcg x once, then 5 mcg per dose if tolerated for 6 to 9 treatments/day.
|
T ½ ~ 20 to 30 min
|
Caution if underlying lung disease or symptomatic hypotension. Bronchospasm
|
One ampule used per treatment session
(20 mcg/ml = 5 mcg dose only!)
Only available through LDD
|
|
Bosentan
(Tracleer® 62.5 mg, 125 mg tablets)
|
Oral tablets. Can be dissolved to make a solution
|
II-IV
|
Initial: 62.5 mg BID x 4 weeks, then increase to 125 mg BID thereafter if tolerated and wt > 40kg.
|
T ½ ~ 5 hours
Metabolized and strong inducer of CYP3A4 and CYP2C9, possibly CYP2C19; Caution with DDI's.
|
CI: Pregnancy and use of cyclosporine or glyburide. Caution with liver disease.
|
REMS for teratogenicity and hepatotoxicity. All patients must enroll in Tracleer Access Program (TAP) Only available through LDD
|
|
Ambrisentan
(Letairis® 5 mg, 10 mg tablets)
|
Oral tablets, should not be split, crushed or chewed.
|
II-III
|
Initial: 5 mg daily, increase to 10 mg daily if tolerated
|
T ½ up to ~ 15 hours
Metabolized by CYP3A4 and CYP2C19, substrate of P-glycoprotien
|
CI: pregnancy and IPF. Caution with anemia, fluid retention, PVOD.
|
REMS for teratogenicity. WRP must enroll in Letairis Education and Access Program (LEAP) Only available through LDD
|
|
Macitentan
(Opsumit® 10 mg tablets)
|
Oral tablets
|
II-III
|
10 mg po daily
|
T½ ~16 hrs (48 hrs for active metabolite)
Metabolized by CYP3A4 and CYP2C19; active metabolite contributes ~40% of activity
|
CI: Pregnancy Caution with anemia, liver disease.
|
REMS for teratogenicity. Must enroll in Opsumit REMS program.
Only available through LDD.
|
|
Sildenafil
(generic, Revatio® 20 mg tablets)
(Revatio® 10 mg/12.5 ml soln for inj.)
|
Oral tablets. Can be extemporaneously compounded. Solution for injection used for NPO
|
Mostly II-III
|
Oral: 20 mg TID
Inj.: 10 mg TID
|
T ½ ~ 4 hours
Metabolized by CYP3A4 and CYP2C9 (minor)
|
CI: use with organic nitrates
Inc mortality risk in peds
Caution with SCD, PVOD
|
Post marketing AE: NAION
|
|
Tadalafil
(Adcirca® 20 mg tablets)
|
Oral tablets
|
II-III
|
40 mg daily
|
T ½ ~ 35 hrs
Metabolized by CYP3A4
|
CI: use with organic nitrates
Caution with SCD, PVOD
|
|
|
Selexipag
(Uptravi® 200 mcg, 400 mcg, 600 mcg, 800 mcg, 1000 mcg, 1200 mcg, 1400 mcg, 1600 mcg)
|
Oral Tablets
|
Mostly II-III
|
Start at 200 mcg BID, titrate by 200mcg BID once weekly to highest tolerated dose (max 1600mcg BID)
|
T ½ ~ 6 to 13.5 hrs for active metabolite
Substrate of 2C8, 3A4, P-gp, BRCP, UGT1A3, UGT2B7, OATP1B1, OATP1B3
|
CI: none
Caution with moderate liver disease (dose adjustment may be necessary). Avoid with severe liver disease
|
Only available through LDD
|
|
Riociguat
(Adempas® 0.5 mg,
1 mg, 1.5 mg, 2 mg, 2.5 mg tablets)
|
Oral Tablets
|
II-III
|
0.5 to 1 mg TID, titrated q2weeks to max 2.5 mg TID
|
T ½ ~12 hrs in PAH pts.
Substrate of P-gp and BCRP, metabolized by CYP-1A1, 3A, 2C8, 2J2.
|
CI: Pregnancy, nitrates, PDE-5i.
Caution with hypotension, PVOD, bleeding, smokers.
|
REMS for teratogenicity. Must enroll in Adempas REMS program.
Only available through LDD.
|
PAH: pulmonary arterial hypertension; FC: functional class; CI/P: contraindications and precautions; CVC: central venous catheter; T ½: half-life; CHF: congestive heart failure; LVD: left ventricular disease; LDD: limited distribution drug; BSI: blood stream infection; IV: intravenous; SubQ: subcutaneous; QID: four times daily; BID: two times daily; wt: weight; REMS: risk evaluation and mitigation strategies; PVOD: pulmonary veno-occlusive disease; SCD: sickle cell disease; NAION: nonarteritic ischemic optic neuropathy; WRP: woman of reproductive potential; NPO: nothing by mouth; DDI: drug-drug interactions
*Next generation formulation of epoprostenol that has improved temperature stability and storage recommendations at specific concentrations. |
The 5th WSPH published an updated recommended treatment algorithm in 2013; this algorithm was developed by a group of international experts based on available evidence from randomized controlled trials (Figure 1).7 Additionally, initial therapy selection recommendations are stratified according to WHO FC and include oral, inhaled, and infused therapies or a combination of two or more of these. In 2015, the European Society of Cardiology (ESC) and the European Respiratory Society (ERS) Joint Task Force published more specific treatment recommendations incorporating patient risk factors and disease severity as well as more specific recommendations for upfront combination therapy.33 In addition to disease severity and prognosis, other factors that may contribute to treatment selection are patient preference, social support and social history, as well as coexisting conditions such as liver or renal impairment.
| Figure 1. 5th World Symposium on PH: 2013 PAH Treatment Algorithm7 |
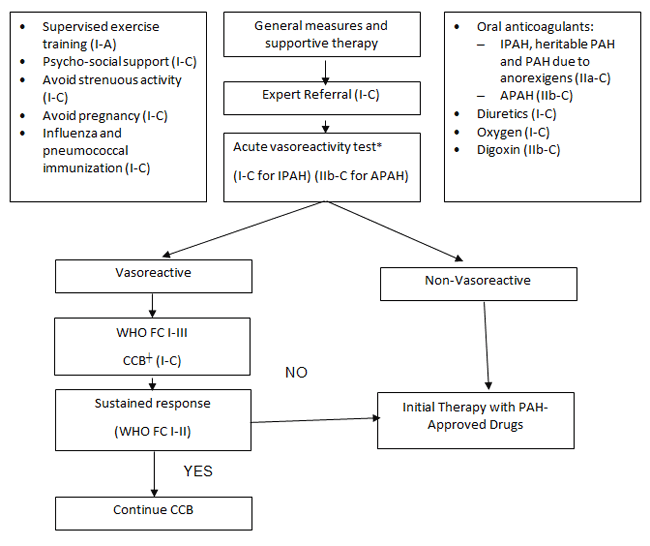 |
*Acute vasodilatory testing should be performed in all patients to determine candidacy for treatment with calcium channel blockers (CCBs). Generally, this is also required for insurance reimbursement.
†Indicated only for patients with a positive response to acute vasoreactivity testing. |
Adjunctive therapy and other health maintenance considerations
Most patients will be assessed for adjunctive therapy with digoxin, oxygen, diuretics, and warfarin.34,35 It should be noted that adjunctive therapy is more supportive in nature and does not affect the underlying pathobiology of the disease and the use of adjunctive therapy, also sometimes referred to as "conventional therapy", should not preclude the use of a PAH targeted therapy. The use of digoxin has fallen out of favor due to its variable inotropic effect, lack of proven benefit, and associated risks. Oxygen can be used to improve hypoxia and prevent vasoconstriction; target saturation is > 90%. Diuretics are utilized by most patients with PAH to treat fluid retention, which may be related to the disease or secondary to some treatments. Anticoagulation can be considered in patients with IPAH as historical studies have demonstrated a mortality benefit in that subgroup, although recent studies question this finding.36
Unless contraindicated, all patients should be vaccinated against pneumococcal infections and annually for influenza.7,35 Women that are capable of reproduction should avoid becoming pregnant due to high reported rates of maternal mortality.37 Of note, it is highly recommended to develop a plan for contraception with these patients.
Calcium channel blocker therapy
A small subset of approximately 10% of patients diagnosed with PAH will have a positive acute vasodilator response during a RHC; a portion of these responders may be treated successfully with calcium channel blockers (CCBs).38,39 It's important to note that patient selection for CCB therapy is crucial and can only be made with evidence of acute vasoreactivity and other hemodynamic measures found during a RHC. CCBs should never be empirically initiated as very few patients are identified as responders, and only about half of those identified will have sustained vasoreactivity and clinical response over time.40 Of the patients that do exhibit a positive response, the preponderance are IPAH and possibly anorexigan-associated PH. The utility of CCB treatment in other WHO Group 1 PAH patients is questionable.41 If CCB therapy is appropriate, other essential considerations are drug selection, dosing, and tolerability. Some CCBs such as verapamil are contraindicated due to the negative inotropic effect on the cardiac vasculature. Of the more commonly used CCBs, including amlodipine, nifedipine, and possibly diltiazem, very high doses that often exceed the labeled maximum dose are generally required. Patients must be monitored frequently for sustained response. Furthermore, because of the inordinately high doses, tolerability issues such as lower extremity edema, hypotension, headaches, and constipation may arise and should be recurrently assessed.
Targeted therapies
Prostacyclin receptor pathway
Prostacyclin is a naturally occurring potent vasodilator that interacts with the prostacyclin receptor on smooth muscle cells in the pulmonary vasculature to promote vasodilation and antiplatelet effects.18 There are multiple PAH medications that target this pathway, either via prostacyclin repletion or through direct interaction with the prostacyclin receptor. These agents differ in temperature stability, pharmacokinetics, and means of drug of delivery that include infused, inhaled and oral routes. Additionally, most of the drugs that affect prostacyclin must be carefully titrated to effect and require extensive patient training and monitoring. Specifically, the infused prostacyclins are typically initiated in a setting of close clinical supervision with experienced personnel due to the profound pharmacologic effects and other risks associated with these therapies. Also fundamental to infused prostacyclin therapy are the risks of rebound pulmonary hypertension resulting in rapid deterioration with infusion interruption and an increased risk of serious bloodstream or catheter-associated infections.
Infused prostacyclins
Epoprostenol sodium
Epoprostenol was the first available PAH-targeted treatment. To date, it is still considered by many experts to be the cornerstone therapy for advanced disease and high-risk patients. There have been numerous studies demonstrating the efficacy of epoprostenol on clinical outcomes in PAH; however, the most noteworthy study was a 12-week, open label, randomized trial of 81 patients comparing intravenous (IV) epoprostenol to conventional therapy.42 This study demonstrated significant improvement in 6MWD, symptoms, and hemodynamics, but most importantly conferred a survival benefit.
Epoprostenol is the most potent of the prostacyclin agents and is administered exclusively by continuous intravenous infusion which requires the placement of a durable tunneled central venous access. Epoprostenol is rapidly hydrolyzed in the blood when exposed to a physiological pH and possesses a very short half-life of six minutes or less.25 Its initiation should only be performed under close clinical monitoring, generally in an inpatient setting. A patient's "dosing weight", or the number of kilograms the patient weighs at the time the prostacyclin infusion is started, is used to calculate the initial dose, mixed concentration, and infusion rate, with titrations typically starting at approximately 2 ng/kg/min. The dose is then titrated to clinical effect by 1 to 2 ng/kg/min increments. Notably, the dosing weight remains fixed for all subsequent titrations and for the duration of therapy.
Epoprostenol is currently available in two formulations, both of which are supplied as a lyophilized powder that requires reconstitution and dilution prior to administration. The type of diluent, frequency of mixing, and the temperature and light exposure stability recommendations differ substantially between the two agents. Flolan® (epoprostenol sodium containing glycine and mannitol) mixed with Flolan® sterile diluent must be protected from light and has a short stability window necessitating refrigeration storage or ice packs during administration.25 Once reconstituted with the standard sterile diluent, the pH is roughly 10.2 to 10.8. Recently, a new high pH sterile diluent was introduced which adjusts the pH to 11.7 to 12.3 to improve the temperature stability.25 Veletri® (epoprostenol sodium containing arginine and mannitol) is a new formulation of epoprostenol with expanded thermal stability and infusion recommendations. It is reconstituted with either sterile water for injection or normal saline and has a pH of approximately 11 to 13. It generally does not require ice packs during administration, but the mixed drug should be refrigerated during storage.26 The two formulations are considered bioequivalent; however, the diluents are not interchangeable so care needs to be taken when switching between the two.43
Possible adverse effects of epoprostenol are listed in Table 5. These side effects are generally class specific so similar management strategies can be used for other prostanoid therapies. Patients who are concomitantly on medications with anti-hypertensive or antiplatelet effects should be closely monitored for additive effects. Additionally, infused prostacyclin is contraindicated for use in patients with congestive heart failure due to left ventricular dysfunction because of the increased risk of mortality.44
| Table 5: Management of Prostacyclin-Related Effects |
| Adverse Effect |
Management Strategy |
| Headache |
OTC analgesics, Tramadol, opiates if severe |
| Diarrhea |
Loperamide, Lomotil, adjust titrations |
| Nausea |
Ondansetron or other anti-emetics, food (oral formulation) |
| Hypotension |
Adjust antihypertensive drugs, diuretics, adjust titrations |
| Dizziness |
Adjust antihypertensive drugs, diuretics, adjust titrations |
| Jaw Pain |
Start first meal with bland food, "exercise jaw" |
| Leg Pain |
Elevate legs, gabapentin, pregabalin, amitriptyline, other pain meds |
| Flushing |
Adjust titrations |
Patients and their caregivers are extensively trained on drug mixing and titrations, pump and supply use, central line care, and emergency preparedness in the event of a pump malfunction, line complication, or other infusion-related problem.
Treprostinil sodium
Treprostinil is a prostacyclin analogue that may also be administered via continuous infusion through a central venous catheter or a less invasive subcutaneous (SQ) method of infusion delivery. The pivotal study of subcutaneous treprostinil was a double blind, 12-week placebo-controlled trial of 470 patients with WHO FC II to IV PAH that evaluated a primary outcome of effect on 6MWD.45The drug successfully improved 6MWD, hemodynamics, and symptoms. There was no significant effect on mortality and SQ site reaction emerged as a frequent adverse effect of treatment. IV treprostinil was found to improve both 6MWD and symptoms as evidenced in the TRUST study, a double-blind placebo-controlled 12-week trial of 44 patients with FC III PAH.46 IV treprostinil is considered to be bioequivalent to the SQ administration method.47
Like epoprostenol, treprostinil infusions are generally initiated during an inpatient admission for close monitoring and titration of the agent. It is initiated at 1.25 ng/kg/min and titrated to response.27 The dose may need to be adjusted for hepatic insufficiency. The IV method of drug delivery also carries the risk of bloodstream infections and catheter-related complications, while SQ administration poses the risk of subcutaneous-related complications such as pain, inflammation, or infection. The half-life of treprostinil is approximately 4 to 6 hours. Additionally, treprostinil is stable at room temperature and is unaffected by exposure to light, which permits the use of microinfusion pumps.27,48 Despite the longer half-life, interruptions must be avoided to prevent complications such as rebound PH and potential death. Treprostinil is infused undiluted through the SQ route; however, when infused intravenously the medication must be diluted in one of three diluent options: normal saline, sterile water for injection, or Flolan® sterile diluent. Many providers prefer using Flolan® sterile diluent because of evidence that it may reduce the risk of infections and also allows for increased storage duration once mixed.49
Oral and inhaled prostacyclins
Treprostinil sodium for inhalation
Treprostinil sodium (Tyvaso®) is also available as an intermittent inhalation formulation using a unique drug- and patient-specific device. It was evaluated in the TRIUMPH 1 trial in which patients on stable background therapy with an endothelin receptor antagonist or phosphodiesterase inhibitor were randomized to placebo or treatment.50 A significant improvement in the primary outcome of 6MWD was identified, secondary outcome improvements in quality-of-life and BNP were also observed. This benefit persisted with an open label 24-month extension study.51
Inhaled treprostinil is typically used in FC III patients who are initiated at a dose of 3 breaths four times daily and titrated to a goal dose of 9 breaths four times daily.23 Doses that exceed 9 breaths four times daily have been evaluated and found to be beneficial and well tolerated for some patients.52 It's important to note that each plastic ampule is multi-dose and provides all of the patient's doses over a 24-hour period. In addition to possible prostacyclin-type adverse effects, patients should be monitored for increased cough, throat irritation, hemoptysis, and pneumonia.
Iloprost
Iloprost was the first available prostacyclin with an intermittent inhalation method of drug delivery. The pivotal trial that resulted in its market approval was AIR. The primary outcome of this study was a composite endpoint of improvement in 6MWD and FC, absence of clinical worsening, and avoidance of death and was found to be significant when compared to placebo.53
Iloprost is administered using the I-neb® AAD® System at a starting dose of 2.5 mcg once, and if tolerated increased to a dose of 5 mcg per dosing session for 6 to 9 total treatment sessions per day.24 The half-life of iloprost is approximately 20 to 30 minutes, necessitating approximately every 2 hour dosing. Each ampule provides one treatment session and ampules are available in two concentrations: 10 mcg/ml which delivers a dose of 2.5 mcg or 5 mcg, and 20 mcg/ml which delivers a dose of only 5 mcg.
Possible adverse effects are related to the vasodilatory effect of prostacyclin therapy and can be similar to other prostacyclins, but may also include increased cough and throat irritation. Caution should be used in patients with underlying lung disease.
Treprostinil diolamine
Treprostinil diolamine is the only available prostacyclin therapy in a tablet in the United States. It was approved in late 2013 based on results from FREEDOM-M, a double-blind placebo-controlled 12-week study of 228 treatment-naïve PAH patients. The primary outcome of improvement in 6MWD proved to be significant.54
Treprostinil diolamine is available as an oral extended release osmotic tablet and typically indicated for functional class I to III patients.32 It is started at a dose of 0.25 mg orally twice daily, or 0.125 mg orally three times daily and titrated every three to four days as tolerated. In general, the three times daily dosing is preferred due to its reduced peak-to-trough ratio and the potential for improved tolerability. Food may increase its bioavailability and therefore is recommended to administer consistently with regards to meals.
Adverse effects for treprostinil are similar to other prostacyclin therapies. However, gastrointestinal effects may be more pronounced and pose a significant challenge for some patients.32 An insoluble shell may be found in the stool and use should be avoided if diverticulosis is present. Liver insufficiency can substantially increase drug exposure and should be avoided with severe hepatic impairment.
IP receptor agonist
Selexipag
Selexipag is a new molecular entity with a novel mechanism that was recently approved by the FDA for the treatment of WHO Group 1 PAH in early 2016. It exerts its effects on the prostacyclin pathway similar to prostacyclins; however, it acts as a selective agonist of the prostacyclin receptors in the pulmonary vasculature. Selexipag is structurally distinct from prostacyclin. GRIPHON was a phase III double blind, randomized, placebo controlled study of patients with PAH who were randomized to selexipag (n=574) or placebo (n=582).55 Most patients enrolled in the study were on background therapy with either monotherapy (47% on ERA or PDE5i) or combination therapy (33% on both ERA and PDE5i) at baseline. Patients were titrated to the highest tolerated dose, ranging from 200mcg to 1600mcg twice daily and observed over time for the primary outcome which was a composite endpoint of death or complication related to PAH. Patients were treated with selexipag for a mean duration of 76 weeks (SD 50.4 weeks) and 71.2 weeks (SD 48.3 weeks) on placebo. In the treatment group, the composite endpoint was reduced by a significant 40% in patients with PAH. This study was important for several reasons in addition to its effect on the primary outcome: it was one of the largest studies in PAH patients to date and it enrolled a substantial number of patients on combination therapy.
Selexipag is administered as an oral tablet and often used in FC II and III patients with PAH.28 It is recommended to start at a dose of 200 mcg twice daily and titrated weekly by increments of 200 mcg twice daily to the highest tolerated dose or a maximum of 1600 mcg twice daily. It's supplied in multiple strengths so patients can be switched to a one tablet twice daily regimen to reduce pill burden once they reach their goal dose. Selexipag is metabolized extensively through the liver and is a substrate of CYP2C8, CYP3A4, and various other enzymes. The dose may need to be adjusted by half in the setting of moderate liver impairment and should be avoided with severe liver disease. The half-life is approximately 6 hours and up to 13.5 hours for its active metabolite. Adverse effects are very similar to other prostacyclin pathway drugs but also include a small but potential risk of hyperthyroidism.
Endothelin Pathway
Endothelin receptor antagonists (ERA)
Endothelin is a potent vasoconstrictor that interacts with ETA and ETB receptors on the pulmonary smooth muscle cells to regulate vascular tone.18 Endothelin activates the ETA receptor resulting in vasoconstriction and cellular proliferation in the small pulmonary arteries which leads to increased pulmonary vascular resistance and vasculature remodeling. Activation of the ETB receptor is thought to have more of a regulatory effect. The primary drug target is antagonism of the ETA receptor to block the effects of endothelin. However, two (bosentan and macitentan) of the three ERAs marketed for the treatment of PAH are dual ETA and ETB receptor blockers while ambrisentan selectively inhibits the ETA receptor. The dual effect does not appear to have an influence on clinical outcomes.
Possible adverse effects of ERAs are generally class specific and include headache, flushing, peripheral edema, and nasal congestion (Table 6). Possible serious adverse effects may include anemia, CHF exacerbation, and risk of birth defects in women of reproductive capabilities who become pregnant. Due to the teratogenic potential of these agents, they carry a black boxed warning and FDA mandated REMS program to reduce the risk of pregnancy.
| Table 6: Management of Oral PAH Targeted Therapy-Related Effects |
| Adverse Effect |
Management Strategy |
| Headache |
OTC analgesics, tramadol, opiates if severe |
| Peripheral Edema |
Add or adjust diuretics, salt and fluid restrictions |
| Anemia |
Periodic CBC monitoring
Reduce dose or discontinue drug |
| Hemorrhagic events Epistaxis (sildenafil) |
Caution with anticoagulants
Monitor for bleeding/bruising |
| Nausea |
Ondansetron or other anti-emetics |
| Hypotension |
Monitor BP in between dose titrations
Adjust antihypertensive drugs, diuretics
Reduce dose or hold titration if needed (riociguat) |
| Dizziness |
Adjust antihypertensive drugs, diuretics |
| Dyspepsia |
PRN OTC agents if infrequent
H2 blocker or PPI |
| Nasal congestion |
Saline nasal spray |
| Teratogenicity |
Obtain negative pregnancy test monthly for women of reproductive age
Contraception mandatory |
| Elevated LFT's |
Monitor LFTs monthly (bosentan) Reduce dose or discontinue drug |
Bosentan
Bosentan was the first available ERA and demonstrated its positive effects in the treatment of PAH in the BREATHE-1 and EARLY studies.56,57 BREATHE-1 evaluated 213 FC II and III PAH patients in a double blind, 16-week, placebo-controlled study and found improvements in 6MWD, symptoms, and a delay in clinical worsening. EARLY assessed 185 FC II patients in a 6-month double-blind placebo-controlled trial that resulted in an improvement in hemodynamics and a delay in clinical worsening. This study provided evidence of improved outcomes with initiation of treatment earlier in the clinical course of the disease.
Bosentan is generally initiated at 62.5 mg twice daily for 1 month and increased to 125 mg twice daily thereafter.21 The dose may need to be adjusted for specific drug interactions (e.g., such as with ritonavir) and patient weight. The use of bosentan may be limited by its drug interactions since it is both metabolized by and a strong inducer of CYP3A4 and CYP2C9. It is also contraindicated for use with cyclosporine and glyburide. Bosentan may cause liver toxicity and has an additional REMS requirement to monitor all patients for hepatotoxicity.
Ambrisentan
The ARIES 1 and ARIES 2 trials were double-blind 12-week placebo-controlled studies in 394 patients with FC II or III PAH that found an improvement in 6MWD and a delay in clinical worsening in patients initiated on 5 mg or 10 mg of study drug compared to placebo.58,59
Ambrisentan is generally started at 5 mg daily for approximately 1 month and increased to 10 mg daily if tolerated. Ambrisentan is contraindicated in patients with idiopathic pulmonary fibrosis due to increased risk of disease progression and must be used cautiously if pulmonary veno-occlusive disease (PVOD) is suspected.60 Ambrisentan has demonstrated safety and efficacy in patients with portopulmonary hypertension and may be a treatment option in this subgroup of patients.61,62
Macitentan
The SERAPHIN study was a milestone in the evolution of PAH investigational trials. This was the first study published that evaluated the long-term effect of any drug on a combined endpoint of reduction in morbidity and mortality events that included death, atrial septostomy, lung transplant, initiation of prostacyclin, or worsening of PAH.63 In this double-blind, event-driven, multicenter study, 742 patients with FC II or III PAH were randomized to receive either macitentan or placebo. The treatment group experienced a significant reduction in risk of morbidity and mortality events, improved 6MWD, and improved symptoms compared to the placebo group. A reduction in hospitalizations in patients with symptomatic PAH was also reported.64
Macitentan is primarily used in FC II and III patients with PAH and dosed at 10 mg once daily.22 It is metabolized through CYP3A4 and CYP2C19 and the active metabolite is responsible for roughly 40% of the drug's activity. Macitentan has increased lipophilicity and slower receptor dissociation compared to other ERAs, which is thought to enhance its pharmacological activity.65
Nitric Oxide Pathway
Nitric oxide is released from endothelial cells and signals the conversion of GTP to cGMP via guanylate cyclase receptors in smooth muscle cells (SMCs) to cause relaxation.18 There are currently two classes of medications that affect the NO pathway and are used for the long-term treatment of PAH. Phosphodiesterase type 5 is the predominant phosphodiesterase in the pulmonary vasculature and is responsible for the inactivation of cGMP. Phosphodiesterase type 5 inhibitors (PDE-5i) block the degradation of cGMP by blocking this enzyme, resulting in improved SMC relaxation. Soluble guanylate cyclase agonists exert a dual mechanism of action along the NO pathway by directly stimulating the gyanylate cyclase receptor independent of NO to increase the production of cGMP, but also assist in stabilizing the endogenous NO-cGMP complex. Inhaled nitric oxide (iNO) also affects this pathway but is generally only used during diagnostic procedures and for more acute, short-term scenarios.
Phosphodiesterase type 5 inhibitors
Sildenafil
SUPER-1 and SUPER-2 evaluated 278 patients with FC 1 to IV PAH in a double-blind placebo-controlled study and 222 patients in an optional 12-month extension period.66,67 Significant improvements in 6MWD, symptoms, and hemodynamics were demonstrated—an effect which persisted for the 1-year extension. Notably, doses up to 80 mg three times daily were found to be beneficial on hemodynamic measures in the extension study; however, only the 20 mg three times daily dose was approved for use based on similar 6MWD outcomes between the treatment groups.
Most patients are initiated on a dose of sildenafil 20 mg three times daily; however, doses up to 80mg three times daily are frequently seen in practice.31 PDE-5i are contraindicated for use in combination with organic nitrate therapies as well as riociguat due to the potentially unsafe effect on blood pressure. Adverse effects may include headache, flushing, nausea, heartburn, priapism, and nasal congestion. Any changes in hearing or vision should be reported to a physician without delay. PDE-5i should be used cautiously in patients with sickle cell anemia due to the increased risk of vaso-occlusive crisis and PVOD associated with an increased risk of pulmonary edema. Specifically, epistaxis may occur with sildenafil, particularly in combination with warfarin.
Tadalafil
PHIRST 1 and PHIRST 2 trials were designed to be double-blind 16-week placebo-controlled studies with an optional 10-month extension period to evaluate the effects of tadalafil 2.5 mg, 10 mg, 20 mg or 40 mg once daily on 6MWD.68,69 Of note, this study allowed background therapy with bosentan. The positive outcomes included improved 6MWD, improved hemodynamics, delay in clinical worsening and improved quality of life. These effects were maintained at the 10-month benchmark and were most pronounced in the tadalafil 40 mg daily group.
The approved dose of tadalafil is 40 mg once daily.30 The dose may need to be adjusted in patients with moderate renal impairment and tadalafil is not recommended in patients with a creatinine clearance of < 30 ml/min or in those who have severe liver impairment (Child Pugh Class C or greater). The possible adverse effects of tadalafil are similar to that of other PDE-5is but also carry a risk of myalgia.
Soluble Guanylate Cyclase Agonists
Riociguat
Riociguat is a first-in-class soluble guanylate cyclase (sGC) agonist and the first treatment for WHO Group 1 PAH to carry an additional approved indication for WHO Group 4 CTEPH. PATENT-1/PATENT-2 was a placebo-controlled, double-blind 12-week study with an opt-in extension period enrollment that evaluated the 443 patients with FC I-IV Group 1 PAH randomized to either riociguat or placebo. CHEST-1/CHEST-2 was a study of similar design that evaluated 261 patients with FC I-IV inoperable, persistent, or recurrent CTEPH.70-73 Both studies found improved 6MWD, improved peripheral vascular resistance (PVR), improved symptoms, and improved N-terminal pro b-type natriuretic peptide (NT-proBNP).
Riociguat is generally initiated at a dose of 0.5 to 1 mg three times daily and titrated no sooner than every 2 weeks by increments of 0.5 mg three times daily to a goal dose of 2.5 mg three times daily.29 Patients must be monitored closely for hypotension and titrations may be adjusted if needed. If three or more days of treatment are missed, the medication must be re-titrated. Riociguat is contraindicated for use in combination with organic nitrates or PDE-5i's, and smokers may require higher-than-average doses. Adverse effects are similar to those of other oral vasodilators, but additionally include higher rates of bleeding as observed in clinical trials. The risk increases when used in combination with warfarin and patients at risk should be carefully monitored. Like the ERAs, riociguat carries a warning for teratogenicity and is subject to REMS requirements to reduce the risk of pregnancy during use.
Combination therapy
Initiating a combination of therapies that target multiple pathways has long been the standard of practice in the treatment of PAH, but until recently evidence to support combination therapy was lacking. Table 7 summarizes studies that have demonstrated positive outcomes when two or more agents were started either sequentially or simultaneously. Of these studies, the AMBITION trial is the only published trial to date designed to look at upfront combination treatment with ambrisentan and tadalafil compared to either agent as monotherapy with an event-driven primary outcome. 57 This was a double-blind placebo-controlled study of 500 FC II and III PAH patients naïve to treatment who were randomized to receive initial combination therapy with ambrisentan plus tadalafil, tadalafil plus placebo, or ambrisentan plus placebo. The combination therapy treatment group was compared to a pooled monotherapy group for changes in the combined primary outcome of death, hospitalization for worsening PAH, progression of PAH, or unsatisfactory clinical response. The primary endpoint occurred significantly less in the combination group than in the monotherapy plus placebo groups (18%, 34%, and 28% respectively). However, there were higher rates of peripheral edema, headache, nasal congestion, and anemia in the combination group compared to monotherapy.
| Table 7 Studies of Combination Therapy50,55,64,67-69,74-77 |
| Study Name |
Positive Outcome |
| PACES (epoprosenol + sildenafil) |
Increased time to clinical worsening |
| STEP (iloprost+bosentan vs. placebo) |
Increased time to clinical worsening |
| TRIUMPH (inhaled treprostinil + sildenafil/bosentan) |
Improved 6MWD |
| PHIRST-1/PHIRST-2 (tadalafil + bosentan vs. placebo) |
Improved and maintained improvement in 6MWD |
| BREATHE-2 (bosentan + epoprostenol) |
Only trend towards clinical improvement |
| COMPASS-2 (bosentan + sildenafil) |
Further improvement in addition of sildenafil to bosentan monotherapy |
| AMBITION (tadalafil + ambrisentan) |
Reduced risk of composite endpoint (death, hospitalization due to PAH, worsening PAH, unsatisfactory long-term response)
Improved 6MWD
Improved N-terminal BNP |
| SERAPHIN (macitentan ± PDE5i or ERA) |
Reduced risk of composite endpoint (death, atrial septostomy, lung transplantation, initiation of treatment infused prostacyclin, or worsening of pulmonary arterial hypertension) |
| GRIPHON (selexipag ± PDE5i or ERA or BOTH) |
Reduced risk of composite endpoint of death or complication related to PAH |
Though the results of AMBITION are promising, there are still many unanswered questions regarding combining agents in the treatment of PAH. For example, data is lacking on which drugs to add, in which order, whether to start simultaneously or add sequentially, safety of potentially additive effects, and the cost effectiveness of dual and triple therapy.
Role of the Specialty Pharmacist in the Care of Patients with PAH
Specialty pharmacists have numerous and multi-faceted opportunities to participate in the management of PAH therapies that span multiple health care settings such as hospitals, ambulatory care specialty clinics, and specialty pharmacies. The specific responsibilities of the specialty pharmacist may vary depending on the practice type but may include education and training, monitoring for drug interactions, medication reconciliation, and ensuring safe-use conditions. For pharmacists who work directly with PAH patients, it is essential to be aware of the unique enrollment and dispensing restrictions that are required to get patients started on many of these therapies. Specialty pharmacists who work in an inpatient setting may participate with policy development, implementing safe-use procedures, coordinating planned admissions for infused prostacyclin initiation, documenting REMS requirements, transitioning therapies, guiding initial dosing and titrations, performing order entry and verification, evaluating patient characteristics for therapy selection, assisting with transitions in care, and assisting with formulary decisions. Those in an ambulatory care clinic have similar responsibilities, but may also help with insurance coverage and drug access through the manufacturers and specialty pharmacies, creating ongoing titrations or other management plans, monitoring medication adherence, managing tolerability issues, coordinating labs for ongoing REMS requirements and evaluating patients for health maintenance such as immunizations. The majority of PAH-targeted therapies are limited distribution drugs and dispensed by a small number of pharmacies nationally. Pharmacists who work in a specialty pharmacy setting generally have specialized training in managing these complex therapies and serve as a liaison between patients and providers, conduct education and training on the drugs and devices, evaluate patients started on therapy for adherence, efficacy, and tolerability, and provide 24/7 pharmacist accessibility for provider or patient questions and for urgent situations (e.g., pump malfunction). The first few months after therapy initiation are critical to long-term success and specialty pharmacists are in a unique position to help facilitate therapeutic decision-making.
Conclusion
Diagnosis and treatment of PAH has come a long way in 20 years, but many obstacles still exist. It is essential for patients to be correctly diagnosed as early in the disease course as possible and for appropriate treatment to be initiated. With new treatment options and more opportunities for combination therapy, treatment plans can be highly individualized based on patient-specific factors. Some PAH therapies are complex in nature and may pose safety concerns. Pharmacists can play an important role in managing these therapies and mitigating possible risks.
REFERENCES
- Farber HW, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med. 2004;351(16):1655-1665. doi:10.1056/NEJMra035488.
- Humbert M, Sitbon O, Chaouat A, et al. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med. 2006;173(9):1023-1030. doi:10.1164/rccm.200510-1668OC.
- Badesch DB, Raskob GE, Elliott CG, et al. Pulmonary arterial hypertension: baseline characteristics from the REVEAL Registry. Chest. 2010;137(2):376-387. doi:10.1378/chest.09-1140.
- Badesch DB, Raskob GE, Elliott CG, et al. Pulmonary arterial hypertension: baseline characteristics from the REVEAL Registry. Chest. 2010;137(2):376-387. doi:10.1378/chest.09-1140.
- D'Alonzo GE, Barst RJ, Ayres SM, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med. 1991;115(5):343-349.
- McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association. J Am Coll Cardiol. 2009;53(17):1573-1619. doi:10.1016/j.jacc.2009.01.004.
- Galiè N, Corris PA, Frost A, et al. Updated treatment algorithm of pulmonary arterial hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D60-72. doi:10.1016/j.jacc.2013.10.031.
- Benza RL, Miller DP, Gomberg-Maitland M, et al. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation. 2010;122(2):164-172. doi:10.1161/CIRCULATIONAHA.109.898122.
- Simonneau G, Gatzoulis MA, Adatia I, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D34-41. doi:10.1016/j.jacc.2013.10.029.
- Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. N Engl J Med. 2009;360(19):1989-2003. doi:10.1056/NEJMra0806188.
- Simonneau G, Gatzoulis MA, Adatia I, et al. Updated Clinical Classification of Pulmonary Hypertension. J Am Coll Cardiol. 2013;62((25 Suppl)):D34-41. doi:10.1016/j.jacc.2013.10.029.
- Hachulla E, Carpentier P, Gressin V. Risk factors for death and the 3-year survival of patients with systemic sclerosis: the French ItinerAIR-Sclerodermie study. Rheumatology. 2009;48:304-308.
- Kieler H, Artama M, Engeland A, et al. Selective serotonin reuptake inhibitors during pregnancy and risk of persistent pulmonary hypertension in the newborn: population based cohort study from the five Nordic countries. BMJ. 2012;344:d8012.
- Hoeper M, Markevych I, Spiekerkoetter E, Welte T, Niedermayer J. Goal-oriented treatment and combination therapy for pulmonary arterial hypertension. Eur Respir J. 2005;26:858-863.
- McLaughlin VV, Gaine SP, Howard LS, et al. Treatment goals of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D73-81. doi:10.1016/j.jacc.2013.10.034.
- McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association. J Am Coll Cardiol. 2009;53(17):1573-1619. doi:10.1016/j.jacc.2009.01.004.
- McLaughlin VV, McGoon MD. Pulmonary arterial hypertension. Circulation. 2006;114(13):1417-1431. doi:10.1161/CIRCULATIONAHA.104.503540.
- Humbert M, Morrell NW, Archer SL, et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43(12 Suppl S):13S-24S. doi:10.1016/j.jacc.2004.02.029.
- Jain S, Ventura H, deBoisblanc B. Pathophysiology of pulmonary arterial hypertension. Seminars in cardiothoracic and vascular anesthesia. Semin Cardiothoracicand Vasc Anesth. 2007;11(2):104-109.
- Letairis [prescribing information]. Foster City, CA. Gilead Sciences. 2015.
- Tracleer [prescribing information]. South San Fransisco, CA. Actelion Pharmaceuticals. 2015.
- Opsumit [prescribing information]. South San Fransisco, CA. Actelion Pharmaceuticals. 2016.
- Tyvaso [prescribing information]. Research Triangle Park, NC. United Therapeutics. 2016.
- Ventavis [prescribing information]. South San Fransisco, CA. Actelion Pharmaceuticals. 2013.
- Flolan [prescribing information]. Research Triangle Park, NC. GlaxoSmithKline. 2016.
- Veletri [prescribing information]. South San Fransisco, CA. Actelion Pharmaceuticals. 2016.
- Remodulin [prescribing information]. Research Triangle Park, NC. United Therapeutics. 2014.
- Uptravi [prescribing information]. South San Fransisco, CA.Actelion Pharmaceuticals. 2016.
- Adempas [prescribing information]. Whippany, NJ.Bayer Healthcare Pharmceuticals. 2014.
- Adcirca [prescribing information]. Indianapolis, IN.Eli Lilly and Company. 2014.
- Revatio [prescribing information]. New York, NY. Pfizer Labs. 2015.
- Orenitram [prescribing information]. Research Triangle Park, NC. United Therapeutics. 2016.
- Lau EMT, Tamura Y, McGoon MD, Sitbon O. The 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: a practical chronicle of progress. Eur Respir J. 2015;46(4):879-882. doi:10.1183/13993003.01177-2015.
- Sauler M, Fares WH, Trow TK. Standard nonspecific therapies in the management of pulmonary arterial hypertension. Clin Chest Med. 2013;34(4):799-810. doi:10.1016/j.ccm.2013.08.013.
- Galiè N, Hoeper MM, Humbert M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J. 2009;30(20):2493-2537. doi:10.1093/eurheartj/ehp297.
- Preston IR, Roberts KE, Miller DP, et al. Effect of Warfarin Treatment on Survival of Patients With Pulmonary Arterial Hypertension (PAH) in the Registry to Evaluate Early and Long-Term PAH Disease Management (REVEAL). Circulation. 2015;132(25):2403-2411. doi:10.1161/CIRCULATIONAHA.115.018435.
- Weiss BM, Zemp L, Seifert B, Hess OM. Outcome of pulmonary vascular disease in pregnancy: a systematic overview from 1978 through 1996. J Am Coll Cardiol. 1998;31(7):1650-1657.
- Rich S, Brundage BH. High-dose calcium channel-blocking therapy for primary pulmonary hypertension: evidence for long-term reduction in pulmonary arterial pressure and regression of right ventricular hypertrophy. Circulation. 1987;76(1):135-141.
- Rich S, Kaufmann E. High dose titration of calcium channel blocking agents for primary pulmonary hypertension: guidelines for short-term drug testing. J Am Coll Cardiol. 1991;18(5):1323-1327.
- Sitbon O, Humbert M, Jaïs X, et al. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation. 2005;111(23):3105-3111. doi:10.1161/CIRCULATIONAHA.104.488486.
- Montani D, Savale L, Natali D. Long-term response to calcium-channel blockers in non-idiopathic pulmonary arterial hypertension. Eur Heart J. 2010;31(15):1898-1907.
- Barst RJ, Rubin LJ, Long WA, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med. 1996;334(5):296-301. doi:10.1056/NEJM199602013340504.
- Greig SL, Scott LJ, Plosker GL. Epoprostenol (Veletri®, Caripul®): a review of its use in patients with pulmonary arterial hypertension. Am J Cardiovasc Drugs Drugs Devices Interv. 2014;14(6):463-470. doi:10.1007/s40256-014-0093-0.
- Califf RM, Adams KF, McKenna WJ, et al. A randomized controlled trial of epoprostenol therapy for severe congestive heart failure: The Flolan International Randomized Survival Trial (FIRST). Am Heart J. 1997;134(1):44-54.
- Simonneau G, Barst RJ, Galie N, et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med. 2002;165(6):800-804. doi:10.1164/ajrccm.165.6.2106079.
- Hiremath J, Thanikachalam S, Parikh K. Exercise improvement and plasma biomarker changes with intravenous treprostinil therapy for pulmonary arterial hypertension: a placebo-controlled trial. J Heart Lung Transplant Off Publ Int Soc Heart Transplant. 2010;29(2):137-149.
- Laliberte K, Arneson C, Jeffs R, Hunt T, Wade M. Pharmacokinetics and steady-state bioequivalence of treprostinil sodium (Remodulin) administered by the intravenous and subcutaneous route to normal volunteers. J Cardiovasc Pharmacol. 2004;44(2):209-214.
- Laliberte K, Jerat S, Tenneson K, Wade M, Zaccardelli D. Continuous low volume delivery of intravenous treprostinil via a central venous catheter with a miniature pump in a conscious dog model. J Vasc Access. 2005;6(4):177-181.
- Zaccardelli D, Phares K, Jeffs R, Doran A, Wade M. Stability and antimicrobial effectiveness of treprostinil sodium in Sterile Diluent for Flolan. Int J Clin Pract. 2010;64(7):885-891. doi:10.1111/j.1742-1241.2009.02307.x.
- McLaughlin VV, Benza RL, Rubin LJ, et al. Addition of inhaled treprostinil to oral therapy for pulmonary arterial hypertension: a randomized controlled clinical trial. J Am Coll Cardiol. 2010;55(18):1915-1922. doi:10.1016/j.jacc.2010.01.027.
- Benza RL, Seeger W, McLaughlin VV, et al. Long-term effects of inhaled treprostinil in patients with pulmonary arterial hypertension: the Treprostinil Sodium Inhalation Used in the Management of Pulmonary Arterial Hypertension (TRIUMPH) study open-label extension. J Heart Lung Transplant Off Publ Int Soc Heart Transplant. 2011;30(12):1327-1333. doi:10.1016/j.healun.2011.08.019.
- Parikh K, Rajagopal S, Fortin T, Tapson V, Poms A. Safety and Tolerability of High-dose Inhaled Treprostinil in Pulmonary Hypertension. J Cardiovasc Pharmacol. 2016;67(4):322-325.
- Olschewski H, Simonneau G, Galiè N, et al. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med. 2002;347(5):322-329. doi:10.1056/NEJMoa020204.
- Jing Z-C, Parikh K, Pulido T, et al. Efficacy and safety of oral treprostinil monotherapy for the treatment of pulmonary arterial hypertension: a randomized, controlled trial. Circulation. 2013;127(5):624-633. doi:10.1161/CIRCULATIONAHA.112.124388.
- Sitbon O, Channick R, Chin KM, et al. Selexipag for the Treatment of Pulmonary Arterial Hypertension. N Engl J Med. 2015;373(26):2522-2533. doi:10.1056/NEJMoa1503184.
- Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002;346(12):896-903. doi:10.1056/NEJMoa012212.
- Galiè N, Rubin L, Hoeper M, et al. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomised controlled trial. Lancet Lond Engl. 2008;371(9630):2093-2100. doi:10.1016/S0140-6736(08)60919-8.
- Galiè N, Olschewski H, Oudiz RJ, et al. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation. 2008;117(23):3010-3019. doi:10.1161/CIRCULATIONAHA.107.742510.
- Oudiz RJ, Galiè N, Olschewski H, et al. Long-term ambrisentan therapy for the treatment of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54(21):1971-1981. doi:10.1016/j.jacc.2009.07.033.
- Raghu G, Behr J, Brown KK, et al. Treatment of idiopathic pulmonary fibrosis with ambrisentan: a parallel, randomized trial. Ann Intern Med. 2013;158(9):641-649. doi:10.7326/0003-4819-158-9-201305070-00003.
- Cartin-Ceba R, Swanson K, Iyer V, Wiesner RH, Krowka MJ. Safety and efficacy of ambrisentan for the treatment of portopulmonary hypertension. Chest. 2011;139(1):109-114. doi:10.1378/chest.10-0574.
- Halank M, Knudsen L, Seyfarth H. Ambrisentan improves exercise capacity and symptoms in patients with portopulmonary hypertension. Z Gastroenterol. 2011;49(9):1258-1262.
- Pulido T, Adzerikho I, Channick RN, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369(9):809-818. doi:10.1056/NEJMoa1213917.
- Channick RN, Delcroix M, Ghofrani H-A, et al. Effect of macitentan on hospitalizations: results from the SERAPHIN trial. JACC Heart Fail. 2015;3(1):1-8. doi:10.1016/j.jchf.2014.07.013.
- Gatfield J, Mueller Grandjean C, Sasse T, Clozel M, Nayler O. Slow receptor dissociation kinetics differentiate macitentan from other endothelin receptor antagonists in pulmonary arterial smooth muscle cells. PloS One. 2012;7(10):e47662. doi:10.1371/journal.pone.0047662.
- Galiè N, Ghofrani HA, Torbicki A, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353(20):2148-2157. doi:10.1056/NEJMoa050010.
- Simonneau G, Rubin LJ, Galiè N, et al. Addition of sildenafil to long-term intravenous epoprostenol therapy in patients with pulmonary arterial hypertension: a randomized trial. Ann Intern Med. 2008;149(8):521-530.
- Galiè N, Brundage BH, Ghofrani HA, et al. Tadalafil therapy for pulmonary arterial hypertension. Circulation. 2009;119(22):2894-2903. doi:10.1161/CIRCULATIONAHA.108.839274.
- Oudiz RJ, Brundage BH, Galiè N, et al. Tadalafil for the treatment of pulmonary arterial hypertension: a double-blind 52-week uncontrolled extension study. J Am Coll Cardiol. 2012;60(8):768-774. doi:10.1016/j.jacc.2012.05.004.
- Ghofrani H-A, D'Armini AM, Grimminger F, et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med. 2013;369(4):319-329. doi:10.1056/NEJMoa1209657.
- Ghofrani H-A, Galiè N, Grimminger F, et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med. 2013;369(4):330-340. doi:10.1056/NEJMoa1209655.
- Rubin LJ, Galiè N, Grimminger F, et al. Riociguat for the treatment of pulmonary arterial hypertension: a long-term extension study (PATENT-2). Eur Respir J. 2015;45(5):1303-1313. doi:10.1183/09031936.00090614.
- Simonneau G, D'Armini AM, Ghofrani H-A, et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension: a long-term extension study (CHEST-2). Eur Respir J. 2015;45(5):1293-1302. doi:10.1183/09031936.00087114.
- Galiè N, Barberà JA, Frost AE, et al. Initial Use of Ambrisentan plus Tadalafil in Pulmonary Arterial Hypertension. N Engl J Med. 2015;373(9):834-844. doi:10.1056/NEJMoa1413687.
- Humbert M, Barst R, Robbins I. Combination of bosentan with epoprostenol in pulmonary arterial hypertension: BREATHE-2. Eur Respir J. 2004;24(3):353-359.
- McLaughlin VV, Oudiz RJ, Frost A, et al. Randomized study of adding inhaled iloprost to existing bosentan in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2006;174(11):1257-1263. doi:10.1164/rccm.200603-358OC.
- McLaughlin V, Channick RN, Ghofrani H-A, et al. Bosentan added to sildenafil therapy in patients with pulmonary arterial hypertension. Eur Respir J. 2015;46(2):405-413. doi:10.1183/13993003.02044-2014.
Back to Top